AZD8835 is a novel, selective, and orally bioavailable inhibitor of the class I (PI3K) catalytic subunit alpha (PIK3CA) PI3Kα and PI3Kδ with IC50s of 6.2 and 5.7 nM, respectively with potential antineoplastic activity. In the PI3K/Akt (protein kinase B)/mammalian target of rapamycin (mTOR) pathway, the PI3K alpha inhibitor AZD8835 specifically binds to and inhibits PIK3CA and its mutant forms. This causes apoptosis in PIK3CA-expressing tumor cells as well as growth inhibition. This medication may be more effective and less harmful than pan-PI3K inhibitors because it targets PIK3CA specifically. The PI3K/Akt/mTOR pathway is frequently dysregulated in solid tumors, which promotes tumor cell growth, survival, and resistance to chemotherapy and radiotherapy.
Physicochemical Properties
| Molecular Formula | C22H31N9O3 |
| Molecular Weight | 469.54 |
| Exact Mass | 469.254 |
| Elemental Analysis | C, 56.28; H, 6.65; N, 26.85; O, 10.22 |
| CAS # | 1620576-64-8 |
| Related CAS # | 1620576-64-8 |
| PubChem CID | 76685059 |
| Appearance | Light yellow to yellow solid powder |
| Density | 1.5±0.1 g/cm3 |
| Boiling Point | 784.0±70.0 °C at 760 mmHg |
| Flash Point | 427.9±35.7 °C |
| Vapour Pressure | 0.0±2.9 mmHg at 25°C |
| Index of Refraction | 1.703 |
| LogP | 3.33 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 10 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 34 |
| Complexity | 685 |
| Defined Atom Stereocenter Count | 0 |
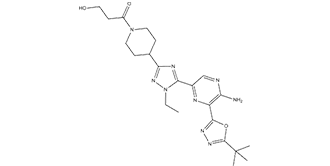
| SMILES | CC(C1OC(C2=C(N)N=CC(C3N(N=C(N=3)C3CCN(C(=O)CCO)CC3)CC)=N2)=NN=1)(C)C |
| InChi Key | ZGRDYKFVDCFJCZ-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C22H31N9O3/c1-5-31-19(26-18(29-31)13-6-9-30(10-7-13)15(33)8-11-32)14-12-24-17(23)16(25-14)20-27-28-21(34-20)22(2,3)4/h12-13,32H,5-11H2,1-4H3,(H2,23,24) |
| Chemical Name | 1-(4-(5-(5-amino-6-(5-(tert-butyl)-1,3,4-oxadiazol-2-yl)pyrazin-2-yl)-1-ethyl-1H-1,2,4-triazol-3-yl)piperidin-1-yl)-3-hydroxypropan-1-one |
| Synonyms | AZD-8835; AZD 8835; AZD8835 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PI3Kδ (IC50 = 5.7 nM); PI3Kα (IC50 = 6.2 nM); PI3Kα-H1047R (IC50 = 5.8 nM); PI3Kα-E545K (IC50 = 6 nM); PI3Kγ (IC50 = 90 nM); PI3Kβ (IC50 = 431 nM)
AZD8835 targets PI3Kα (IC50 = 0.02 μM), PI3Kδ (IC50 = 0.03 μM) [1] AZD8835 shows moderate activity against PI3Kβ (IC50 = 0.3 μM) and weak activity against PI3Kγ (IC50 = 2.5 μM) [2] |
| ln Vitro |
AZD8835 is a potent inhibitor of PI3Kα (wild type, E545K and H1047R mutations) and PI3Kδ with excellent selectivity vs. PI3Kβ, PI3Kγ and an excellent general kinase selectivity. AZD8835 is a potent inhibitor of p-Akt in cells sensitive to PI3Kα inhibition (IC50=0.057 μM in PIK3CA mutant human breast ductal carcinoma BT474 cell line) and in cells sensitive to PI3Kδ inhibition (IC50=0.049 μM in JeKo-1 B cell line), but not to cells sensitive to PI3Kβ inhibition (IC50=3.5 μM in PTEN null breast adenocarcinoma MDA-MB-468 cell line) or PI3Kγ inhibition (IC50=0.53 μM in monocytic RAW264 cell line)[2]. In human cancer cell lines with PI3K pathway activation (MCF-7, BT474, HCT116, U87MG), AZD8835 (0.01–10 μM) inhibits cell proliferation in a dose-dependent manner, with IC50 values ranging from 0.08 to 0.9 μM. MCF-7 (breast cancer) and HCT116 (colon cancer) cells show highest sensitivity (IC50 = 0.08–0.12 μM) [1] - It blocks PI3K-AKT signaling: reduces phosphorylation of AKT (Ser473), GSK3β (Ser9), and S6 ribosomal protein (Ser235/236) in MCF-7 cells (Western blot), with maximal inhibition at 0.5 μM [1] - In BT474 cells, AZD8835 (0.1–1 μM) induces G1 cell cycle arrest (62% of cells in G1 vs. 45% control) and apoptosis (Annexin V-FITC/PI staining shows apoptotic rate ~42% at 0.5 μM) [1] - It exhibits high selectivity for class I PI3Ks: no significant inhibition of 40 unrelated kinases (e.g., mTOR, ERK1/2, JAK2, CDK2) at 10 μM (kinase selectivity panel assay) [2] - In human peripheral blood mononuclear cells (PBMCs), AZD8835 (0.05–1 μM) inhibits LPS-induced TNF-α and IL-6 production (IC50 = 0.1 μM and 0.15 μM, respectively) without affecting cell viability [2] |
| ln Vivo |
AZD8835 exhibits high metabolic stability and suitable physical properties for oral administration, and it has antitumor efficacy in corresponding breast cancer xenograft models when dosed continuously[1][2]. In a subcutaneous xenograft model of breast cancer (MCF-7 cells in nude mice), oral administration of AZD8835 (10 mg/kg/day) for 21 days inhibits tumor growth by ~70% compared to vehicle control. Tumor tissues show reduced p-AKT, p-S6, and Ki-67 expression (immunohistochemistry), and increased cleaved caspase-3 levels (Western blot) [1] - In a colon cancer xenograft model (HCT116 cells in nude mice), oral AZD8835 (15 mg/kg/day) for 28 days prolongs median survival from 32 days (control) to 56 days. It also reduces tumor angiogenesis (CD31 staining shows ~55% reduction in microvessel density) [1] - In a rat model of acute inflammation (carrageenan-induced paw edema), oral AZD8835 (5 mg/kg) reduces paw swelling by ~45% at 4 hours post-administration, with serum TNF-α and IL-6 levels reduced by ~50% [2] |
| Enzyme Assay |
The selectivity profile of AZD8835 (Compound 25) among the class I PI3K isoforms is tested in enzyme and cell based assays. At the enzyme level, AZD8835 is a potent mixed inhibitor of PI3Kα (IC506.2 nM) and PI3Kδ (IC505.7 nM), with selectivity against PI3Kβ (IC50431 nM) and PI3Kγ (IC5090 nM). AZD8835 is also a potent inhibitor of the commonly occurring PI3Kα mutants, PI3Kα- E545K (IC506 nM) and PI3Kα-H1047R (IC505.8 nM). In cell-based assays assessing the ability to inhibit Akt phosphorylation, AZD8835 is a potent inhibitor in cells sensitive to PI3Kα inhibition (IC5057 nM inPIK3CAmutant human breast ductal carcinoma BT474 cell line) and in cells sensitive to PI3Kδ inhibition (IC5049 nM in Jeko-1 B cell line, but not to cells sensitive to PI3Kβ inhibition (IC503.5 μM in PTEN null breast adenocarcinoma MDA-MB-468 cells) or to PI3Kγ inhibition (IC50530 nM in monocytic RAW264 cell line). Class I PI3K kinase activity assay: Recombinant human PI3Kα (p110α/p85α), PI3Kβ (p110β/p85α), PI3Kγ (p110γ/p101), and PI3Kδ (p110δ/p85α) were each incubated with phosphatidylinositol substrate, ATP, and reaction buffer (20 mM Tris-HCl pH 7.5, 10 mM MgCl2, 1 mM DTT) at 30°C for 60 minutes. AZD8835 was added at concentrations ranging from 0.001–10 μM. Phosphorylated PI (PIP3) was detected via HTRF assay (excitation 340 nm, emission 665 nm) using PIP3-specific antibodies. IC50 values were calculated by nonlinear regression of dose-response curves [1,2] - Kinase selectivity assay: AZD8835 (10 μM) was incubated with 40 purified kinases (including mTOR, ERK1/2, JAK2, CDK2, EGFR) and respective substrates/ATP under standard kinase assay conditions. Kinase activity was measured via radiometric or fluorescence-based assays, and inhibition percentage was calculated to confirm selectivity for class I PI3Ks [2] |
| Cell Assay |
BT474, MCF7, or T47D cells are seeded in 384-well plates at a density of 500–2,000 cells per well and incubated overnight. Over the course of several days, cells are dosed with a compound or compounds, and cell confluency is measured every four hours. Cancer cell proliferation and signaling assay: MCF-7/BT474/HCT116 cells (5×10³ per well) were seeded in 96-well plates, treated with AZD8835 (0.01–10 μM) for 48 hours. Cell viability was measured by CCK-8 assay to determine IC50. For signaling analysis, cells were treated with the drug (0.05–1 μM) for 24 hours, lysed, and Western blot was performed to detect p-AKT, AKT, p-GSK3β, GSK3β, p-S6, S6, and GAPDH [1] - Cell cycle and apoptosis assay: BT474 cells (1×10⁵ per well) were seeded in 6-well plates, treated with AZD8835 (0.1–1 μM) for 24 hours. Cell cycle was analyzed by PI staining and flow cytometry; apoptosis was detected by Annexin V-FITC/PI staining and flow cytometry [1] - PBMC cytokine production assay: Human PBMCs were isolated, seeded in 24-well plates, pretreated with AZD8835 (0.05–1 μM) for 1 hour, then stimulated with LPS (1 μg/mL) for 24 hours. Supernatants were collected, and TNF-α/IL-6 levels were quantified by ELISA [2] |
| Animal Protocol |
CD1 mice 0.1 mL/10 g mouse oral administration Breast cancer subcutaneous xenograft model: Nude mice (4-week-old, female) were subcutaneously injected with MCF-7 cells (5×10⁶ cells/mouse) into the right flank. When tumors reached ~100 mm³, mice were randomized into control (n = 6) and AZD8835 treatment (n = 6) groups. The drug was dissolved in 0.5% carboxymethylcellulose (CMC) + 0.1% Tween 80, administered orally at 10 mg/kg once daily for 21 days. Tumor volume (length×width²/2) and body weight were measured every 3 days; tumors were excised for immunohistochemistry and Western blot [1] - Colon cancer xenograft model: Nude mice (4-week-old, male) were subcutaneously injected with HCT116 cells (5×10⁶ cells/mouse). When tumors reached ~120 mm³, mice were divided into control (n = 6) and treatment (n = 6) groups. AZD8835 was administered orally at 15 mg/kg once daily for 28 days. Survival time was recorded, and tumor tissues were analyzed for angiogenesis (CD31 staining) [1] - Acute inflammation rat model: Male Sprague-Dawley rats (250–300 g) were injected with carrageenan (1% w/v, 0.1 mL) into the hind paw to induce edema. AZD8835 was dissolved in DMSO (5%) + saline (95%), administered orally at 5 mg/kg 1 hour post-carrageenan injection. Paw volume was measured at 0, 2, 4, 6 hours post-drug administration; serum was collected for cytokine analysis [2] - Pharmacokinetic study: Male Sprague-Dawley rats (250–300 g) and beagle dogs (8–10 kg) were administered AZD8835 via oral gavage (10 mg/kg) or intravenous injection (2 mg/kg). Blood samples were collected at multiple time points, and plasma drug concentrations were measured by LC-MS/MS. Pharmacokinetic parameters (Cmax, AUC, t1/2, F) were calculated using non-compartmental analysis [2] |
| ADME/Pharmacokinetics |
Oral bioavailability: 70% in rats, 75% in dogs [2] - Plasma half-life (t1/2): 4.2 hours in rats, 8.5 hours in dogs [2] - Plasma protein binding rate: 92% in human plasma, 90% in rat plasma, 93% in dog plasma (equilibrium dialysis assay) [2] - Tissue distribution: In rats, highest concentrations in liver (3.0-fold vs. plasma), kidney (2.7-fold vs. plasma), and tumor tissues (2.3-fold vs. plasma); minimal penetration into the central nervous system (<1.5% of plasma concentration) [2] - Metabolism: Primarily metabolized via hepatic CYP3A4 and CYP2C9-mediated oxidation; major metabolites are hydroxylated derivatives (non-active) [2] - Excretion: In rats, 57% excreted in feces, 33% in urine within 72 hours post-administration [2] |
| Toxicity/Toxicokinetics |
In vitro toxicity: AZD8835 at concentrations up to 10 μM shows no significant cytotoxicity to normal human mammary epithelial cells (HMEC) or PBMCs (cell viability >85% vs. control) [1,2] - Acute toxicity: LD50 > 2000 mg/kg in rats and mice (oral administration); no mortality or severe toxic symptoms (lethargy, convulsions) observed at doses up to 2000 mg/kg [1] - Repeat-dose toxicity: In a 28-day study in rats (oral doses of 5, 15, 50 mg/kg/day), the drug was well-tolerated. Minimal gastrointestinal discomfort (transient soft stools) was observed only at 50 mg/kg; no changes in body weight, hematological parameters, or serum chemistry (ALT, AST, BUN, creatinine) were detected. Histological examination of major organs revealed no abnormal lesions [1] - Drug-drug interaction potential: AZD8835 does not inhibit or induce major CYP450 enzymes (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4) at therapeutic concentrations [2] |
| References |
[1]. Mol Cancer Ther . 2016 May;15(5):877-89. [2]. Bioorg Med Chem Lett . 2015 Nov 15;25(22):5155-62. |
| Additional Infomation |
PI3Kalpha Inhibitor AZD8835 is an orally bioavailable inhibitor of the class I phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) catalytic subunit alpha (PIK3CA), with potential antineoplastic activity. PI3K alpha inhibitor AZD8835 selectively binds to and inhibits PIK3CA and its mutated forms, in the PI3K/Akt (protein kinase B) /mammalian target of rapamycin (mTOR) pathway. This results in both apoptosis and growth inhibition in PIK3CA-expressing tumor cells. By specifically targeting PIK3CA, this agent may be more efficacious and less toxic than pan-PI3K inhibitors. Dysregulation of the PI3K/Akt/mTOR pathway is often found in solid tumors and results in the promotion of tumor cell growth, survival, and resistance to chemo- and radio-therapy. PIK3CA, one of the most frequently mutated oncogenes, encodes the p110-alpha catalytic subunit of the class I PI3K. AZD8835 is a potent, orally bioavailable, and selective class I PI3K inhibitor with preferential activity against PI3Kα and PI3Kδ [1,2] - Its mechanism of action involves inhibiting PI3K-mediated AKT activation, leading to cell cycle arrest and apoptosis in cancer cells with dysregulated PI3K pathway, and suppressing pro-inflammatory cytokine production in immune cells [1,2] - It shows preclinical efficacy in breast cancer, colon cancer, and inflammatory models, supporting potential therapeutic applications in solid tumors and inflammatory disorders [1,2] - Favorable oral bioavailability, metabolic stability, and low toxicity profile make it suitable for oral administration in clinical settings [2] - It has been evaluated in preclinical studies for cancer therapy, with a focus on tumors dependent on PI3Kα/δ signaling [1] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~93 mg/mL (~198.1 mM) Water: <1 mg/mL Ethanol: <1 mg/mL |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 0.83 mg/mL (1.77 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 8.3 mg/mL clear DMSO stock solution to 400 μL of PEG300 and mix evenly; then add 50 μL of Tween-80 to the above solution and mix evenly; then add 450 μL of normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 0.83 mg/mL (1.77 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 8.3 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 0.83 mg/mL (1.77 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 8.3 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1297 mL | 10.6487 mL | 21.2974 mL | |
| 5 mM | 0.4259 mL | 2.1297 mL | 4.2595 mL | |
| 10 mM | 0.2130 mL | 1.0649 mL | 2.1297 mL |