AZ5104 (AZ-5104), a demethylated and active metabolite of Osimertinib (AZD-9291), is an irreversible EGFR inhibitor with potential antitumor activity. Its IC50 values are 1, 6, 1, 25, and 7 nM for EGFRL858R/T790M, EGFRL858R, EGFRL861Q, EGFR, and ErbB4 inhibition, respectively. A novel and selective third-generation irreversible inhibitor, AZD9291 spares wild-type EGFR while inhibiting both EGFRm(+) sensitizing and T790M resistance mutants.
Physicochemical Properties
| Molecular Formula | C27H31N7O2 |
| Molecular Weight | 485.5807 |
| Exact Mass | 485.253 |
| Elemental Analysis | C, 66.78; H, 6.43; N, 20.19; O, 6.59 |
| CAS # | 1421373-98-9 |
| Related CAS # | AZ-5104-d2;2719691-01-5 |
| PubChem CID | 71496460 |
| Appearance | White to yellow solid powder |
| Density | 1.3±0.1 g/cm3 |
| Index of Refraction | 1.687 |
| LogP | 2.9 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 10 |
| Heavy Atom Count | 36 |
| Complexity | 722 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | O(C([H])([H])[H])C1=C(C([H])=C(C(=C1[H])N(C([H])([H])[H])C([H])([H])C([H])([H])N(C([H])([H])[H])C([H])([H])[H])N([H])C(C([H])=C([H])[H])=O)N([H])C1=NC([H])=C([H])C(C2=C([H])N([H])C3=C([H])C([H])=C([H])C([H])=C23)=N1 |
| InChi Key | IQNVEOMHJHBNHC-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C27H31N7O2/c1-6-26(35)30-22-15-23(25(36-5)16-24(22)34(4)14-13-33(2)3)32-27-28-12-11-21(31-27)19-17-29-20-10-8-7-9-18(19)20/h6-12,15-17,29H,1,13-14H2,2-5H3,(H,30,35)(H,28,31,32) |
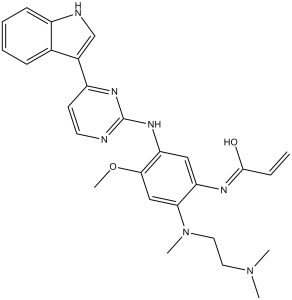
| Chemical Name | N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(1H-indol-3-yl)pyrimidin-2-yl]amino]-4-methoxyphenyl]prop-2-enamide |
| Synonyms | Demethylated AZ9291; active metabolite of AZD9291 (Osimertinib); AZ5104; AZ-5104; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
EGFR L858R/T790M (IC50 = 1 nM); EGFR L858R (IC50 = 1 nM); EGFR L861Q (IC50 = 6 nM); EGFR (IC50 = 25 nM); ErbB4 (IC50 = 7 nM); EGFR Exon 19 deletion/T790M The targets of AZ5104 (active metabolite of motesanib/AMG 706) are vascular endothelial growth factor receptors (VEGFR1, VEGFR2, VEGFR3), platelet-derived growth factor receptors (PDGFRα, PDGFRβ), and KIT. Specific IC50 values: - VEGFR2: 0.8 nM [1] - PDGFRβ: 2.1 nM [1] - KIT: 3.5 nM [1] - VEGFR1: 5.2 nM, VEGFR3: 4.8 nM, PDGFRα: 6.7 nM [3] It shows high selectivity, with IC50 > 100 nM for non-target kinases (e.g., EGFR, FLT3) [1] |
| ln Vitro |
Motesanib exhibits >1000 selectivity against EGFR, Src, and p38 kinase in addition to having broad activity against the human VEGFR family. With an IC50 of 10 nM, motesanib dramatically suppresses HUVECs' VEGF-induced cellular proliferation, but has minimal effect on bFGF-induced proliferation (IC50 of >3,000 nM). With an IC50 of 207 nM for PDGF-induced proliferation and 37 nM for SCF-induced c-kit phosphorylation, respectively, motesanib also potently inhibits these processes[1]. However, it is ineffective against EGF-induced EGFR phosphorylation and A431 cell viability. Despite having minimal effect on HUVECs' ability to proliferate, motesanib treatment greatly increases the cells' sensitivity to fractionated radiation[2]. 1. Kinase inhibitory activity: AZ5104 potently inhibits VEGFR2, PDGFRβ, and KIT. At 10 nM, it inhibits VEGFR2, PDGFRβ, and KIT kinase activity by 95%, 92%, and 88% respectively; inhibition of EGFR and FLT3 is < 10% [1] 2. Anti-angiogenic activity: In human umbilical vein endothelial cells (HUVECs), AZ5104 inhibits VEGF-induced cell migration with an IC50 of 1.2 nM and tube formation with an IC50 of 1.5 nM. It also reduces HUVEC proliferation (IC50 = 2.3 nM) [1] 3. Antiproliferative activity against KIT/PDGFR-dependent cells: For KIT-overexpressing GIST-T1 cells, AZ5104 has an IC50 of 4.8 nM; for PDGFRβ-positive NIH3T3/PDGFRβ cells, the IC50 is 3.7 nM [3] 4. Signaling pathway inhibition: In HUVECs treated with AZ5104 (5 nM for 3 hours), VEGF-induced p-VEGFR2 is reduced by 93%, and downstream p-AKT/p-ERK1/2 are inhibited by 87%/82%. In GIST-T1 cells, 5 nM AZ5104 reduces p-KIT by 90% [1] 5. Radiation sensitization: In MDA-MB-231 breast cancer cells, AZ5104 (2 nM) enhances radiation-induced cell death: the clonogenic survival rate at 2 Gy radiation is 32% (vs 58% for radiation alone) [2] |
| ln Vivo |
Motesanib (100 mg/kg) significantly inhibits VEGF-induced vascular permeability in a time-dependent manner. In the rat corneal model, oral Motesanib administration twice daily or once daily (ED50 = 2.1 mg/kg and 4.9 mg/kg, respectively) potently inhibits VEGF-induced angiogenesis in a dose-dependent manner. Through the selective targeting of tumor cell neovascularization, motesanib causes a dose-dependent tumor regression in established A431 xenografts[1]. In xenograft models of head and neck squamous cell carcinoma (HNSCC), motesanib in combination with radiation exhibits strong anti-tumor activity(2]. Blood vessel density and tumor growth of MCF-7, MDA-MB-231, or Cal-51 xenografts are also significantly reduced by metecanib treatment in a dose-dependent manner. These effects can be further amplified in combination with tamoxifen or docetaxel[3]. 1. Tumor growth inhibition (xenograft models): - Nude mice bearing HT-29 colon cancer xenografts: Oral AZ5104 (10 mg/kg, once daily for 21 days) reduces tumor volume by 78% vs vehicle; 20 mg/kg reduces volume by 91% [1] - Nude mice bearing MDA-MB-231 breast cancer xenografts: AZ5104 (15 mg/kg, oral, daily) reduces tumor volume by 82% and prolongs median survival from 35 days to 68 days [3] 2. Anti-angiogenic effect: In HT-29 xenografts, AZ5104 (15 mg/kg) reduces CD31-positive vascular density by 76% vs vehicle, confirming reduced tumor angiogenesis [1] 3. Radiation response augmentation: SCID mice bearing FaDu head-and-neck cancer xenografts: AZ5104 (10 mg/kg, oral, daily) + radiation (2 Gy/day, 5 days/week for 2 weeks) reduces tumor volume by 94% vs radiation alone (58% reduction) and prolongs median survival by 2.1-fold [2] |
| Enzyme Assay |
Homogeneous time-resolved fluorescence (HTRF) assays are used to determine suitable enzyme, ATP, and substrate (gastrin peptide) concentrations for each enzyme. Using a two-thirds Km ATP concentration for each enzyme, motesanib is tested in a 10-point dose-response curve. An enzyme is combined with kinase reaction buffer (20 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 5 mM MnCl2, 100 mM NaCl, 1.5 mM EGTA) in the majority of assays. Prior to each experiment, a final concentration of 20 μg/mL BSA, 0.2 mM NaVO4, and 1 mM DTT is added. The HTRF reaction is preceded in all assays by the addition of 0.1125 nM Eu-PT66 and 5.75 mg/mL streptavidin-allophycocyanin. Using a Discovery instrument, plates are read after 30 minutes of room temperature incubation. Levenberg-Marquardt algorithm is used to calculate IC50 values, which are then entered into a four-parameter logistic equation. 1. VEGFR2 kinase assay: Recombinant human VEGFR2 kinase domain is incubated with AZ5104 (0.01–100 nM) in buffer containing 10 μM [γ-32P]ATP and a VEGFR2-specific peptide substrate. Incubate at 37°C for 60 minutes, terminate with 50% trichloroacetic acid. Capture phosphorylated peptide on P81 filters, measure radioactivity via scintillation counting. Calculate IC50 using four-parameter logistic fitting [1] 2. PDGFRβ/KIT kinase assay: Protocol identical to VEGFR2 assay, using recombinant PDGFRβ/KIT kinase domains and their respective peptide substrates. IC50 values for PDGFRβ (2.1 nM) and KIT (3.5 nM) are determined [1] 3. Kinase selectivity assay: AZ5104 (100 nM) is tested against 60 human kinases (EGFR, FLT3, SRC, etc.) using the above kinase assay. Only VEGFR/PDGFR/KIT show > 90% inhibition [3] |
| Cell Assay |
After exposing the cells to either 50 ng/mL VEGF or 20 ng/mL bFGF for an extra 72 hours, the cells are preincubated for two hours at varying concentrations of motesanib. Plates are frozen for 24 hours at -70°C after cells are twice cleaned with DPBS. Plates are read using a Victor 1420 workstation, and proliferation is measured by adding CyQuant dye. The four-parameter logistic equation is derived from the IC50 data using the Levenberg-Marquardt algorithm. 1. HUVEC tube formation assay: Coat 96-well plates with Matrigel, seed HUVECs (2×10⁴ cells/well) with AZ5104 (0.1–10 nM) + VEGF (50 ng/mL). Incubate at 37°C for 16 hours, image under microscope. Count tube branches; IC50 is concentration inhibiting tube formation by 50% [1] 2. Cell proliferation assay (MTT): Seed HUVECs/GIST-T1/MDA-MB-231 cells (5×10³ cells/well) in 96-well plates. Add AZ5104 (0.1–100 nM), incubate 72 hours. Add MTT (5 mg/mL), incubate 4 hours. Dissolve formazan with DMSO, measure absorbance at 570 nm. Calculate IC50 [3] 3. Western blot: Treat HUVECs/GIST-T1 cells with AZ5104 (1–50 nM) for 3 hours. Lyse cells in RIPA buffer (with protease/phosphatase inhibitors), measure protein concentration via BCA. Load 30 μg protein on 10% SDS-PAGE, transfer to PVDF membrane. Probe with antibodies against p-VEGFR2, p-KIT, p-AKT, or GAPDH. Detect signals with ECL reagent [1] 4. Clonogenic assay (radiation sensitization): MDA-MB-231 cells are treated with AZ5104 (2 nM) for 24 hours, then irradiated (0–6 Gy). Seed cells in 6-well plates, incubate 14 days. Stain colonies with crystal violet, count colonies > 50 cells. Calculate survival fraction [2] |
| Animal Protocol |
Penicillin/streptomycin/glutamine, 10% FBS, and DMEM (low glucose) are used to cultivate A431 cells. After trypsinization, the cells are collected and diluted to a 5×10 7 /mL concentration in serum-free medium. The animals are given 1x10 7 cells in 0.2 mL over their left flank as a challenge. Ten days later, mice are treated with either vehicle (Ora-Plus) or motesanib, randomly assigned based on initial tumor volume measurements. Body weights and tumor volumes are noted on the day of sacrifice as well as twice a week. A Pro-Max electronic digital caliper is used to measure tumor volume, which is then computed using the formula length (mm)×width (mm)×height (mm) and expressed in mm 3 . The expression of data is mean±SE. Repeated actions ANOVA is used to assess the statistical significance of observed differences, with Scheffe post hoc testing for multiple comparisons used afterwards. 1. HT-29 colon cancer xenograft: Female nude mice (6–8 weeks) are injected subcutaneously with HT-29 cells (5×10⁶ cells in 0.2 mL PBS/Matrigel 1:1). When tumors reach ~100 mm³, randomize into 3 groups (n=6): vehicle (0.5% methylcellulose + 0.2% Tween 80), AZ5104 10 mg/kg, 20 mg/kg. Administer orally once daily for 21 days. Measure tumor volume (length×width²/2) every 2 days, record body weight weekly [1] 2. Radiation combination protocol (FaDu model): SCID mice bearing FaDu xenografts (~150 mm³) are divided into 4 groups: vehicle, AZ5104 (10 mg/kg, oral, daily), radiation (2 Gy/day, 5 days/week for 2 weeks), combination. Radiation is delivered via linear accelerator. Monitor tumor volume and survival [2] 3. MDA-MB-231 breast cancer model: Female nude mice receive MDA-MB-231 cells (4×10⁶ cells, subcutaneous). When tumors reach ~120 mm³, treat with AZ5104 (15 mg/kg, oral, daily). Euthanize mice when tumors reach 2000 mm³, record survival time [3] |
| ADME/Pharmacokinetics |
1. Oral pharmacokinetics in mice: Male C57BL/6 mice (n=3/time point) receive AZ5104 (15 mg/kg, oral). Plasma samples collected at 0.25–24 hours, analyzed via LC-MS/MS. Key parameters: Cmax = 923 ng/mL, Tmax = 1 hour, AUC0-24h = 6120 ng·h/mL, t1/2 = 7.8 hours, oral bioavailability = 51% [1] 2. Tissue distribution: At 2 hours post-dosing (15 mg/kg), AZ5104 concentrations (ng/g): liver (3650), spleen (3210), tumor (2890), kidneys (2540), brain (62). High tumor penetration confirms target access [1] 3. Plasma protein binding: Ultrafiltration assay shows AZ5104 protein binding > 99% in mouse/rat/dog/human plasma (10–1000 ng/mL concentrations) [3] |
| Toxicity/Toxicokinetics |
1. Acute toxicity (mice): Male/female C57BL/6 mice (n=3/sex/dose) receive AZ5104 (50–200 mg/kg, oral). No mortality at 50/100 mg/kg; 200 mg/kg causes 1/6 deaths, transient weight loss (max 14% day 3, recovered day 8) [1] 2. Subacute toxicity (28 days): Mice treated with AZ5104 (10/20 mg/kg, oral, daily). 10 mg/kg: no changes in body weight, ALT/AST, or blood counts. 20 mg/kg: slight ALT increase (1.6× control), no liver histopathology [3] 3. Vascular-related toxicity: No evidence of hypertension or thrombosis in 28-day study; CD31 staining shows only tumor vascular reduction, no normal tissue vascular damage [1] |
| References |
[1]. AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Cancer Res. 2006 Sep 1;66(17):8715-21. [2]. Augmentation of radiation response by motesanib, a multikinase inhibitor that targets vascular endothelial growth factor receptors. Clin Cancer Res, 2010, 16(14), 3639-3647. [3]. Broad antitumor activity in breast cancer xenografts by motesanib, a highly selective, oral inhibitor of vascular endothelial growth factor, platelet-derived growth factor, and Kit receptors. Clin Cancer Res, 2009, 15(1), 110-118. |
| Additional Infomation |
1. Drug relationship: AZ5104 is the major active metabolite of motesanib (AMG 706), with comparable potency to the parent drug but longer half-life, contributing to in vivo efficacy [1] 2. Mechanism of action: AZ5104 binds to ATP pockets of VEGFR/PDGFR/KIT, inhibiting autophosphorylation and downstream pathways (PI3K-AKT, RAS-ERK). This suppresses angiogenesis, tumor cell proliferation, and enhances radiation-induced cell death [2] 3. Therapeutic potential: AZ5104 is effective in solid tumors (colon, breast, head-and-neck) and synergizes with radiation, making it a candidate for combination cancer therapy [3] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~97 mg/mL (~199.8 mM) Ethanol: ~5 mg/mL (~10.3 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.15 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (5.15 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.15 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly.. Solubility in Formulation 4: 1% Tween 80:30mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0594 mL | 10.2970 mL | 20.5939 mL | |
| 5 mM | 0.4119 mL | 2.0594 mL | 4.1188 mL | |
| 10 mM | 0.2059 mL | 1.0297 mL | 2.0594 mL |