Physicochemical Properties
| Molecular Formula | C11H5F2NO4S |
| Molecular Weight | 285.2235 |
| Exact Mass | 284.99 |
| Elemental Analysis | C, 46.32; H, 1.77; F, 13.32; N, 4.91; O, 22.44; S, 11.24 |
| CAS # | 648449-76-7 |
| Related CAS # | 648449-76-7 |
| PubChem CID | 11492951 |
| Appearance | White to off-white solid powder |
| Density | 1.7±0.1 g/cm3 |
| Index of Refraction | 1.658 |
| LogP | 2.63 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 1 |
| Heavy Atom Count | 19 |
| Complexity | 468 |
| Defined Atom Stereocenter Count | 0 |
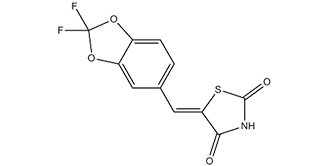
| SMILES | S1C(N([H])C(/C/1=C(\[H])/C1C([H])=C([H])C2=C(C=1[H])OC(O2)(F)F)=O)=O |
| InChi Key | SRLVNYDXMUGOFI-XBXARRHUSA-N |
| InChi Code | InChI=1S/C11H5F2NO4S/c12-11(13)17-6-2-1-5(3-7(6)18-11)4-8-9(15)14-10(16)19-8/h1-4H,(H,14,15,16)/b8-4+ |
| Chemical Name | (5E)-5-[(2,2-difluoro-1,3-benzodioxol-5-yl)methylidene]-1,3-thiazolidine-2,4-dione |
| Synonyms | AS604850; AS 604850; AS-604850 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PI3Kγ (IC50 = 0.25 μM); PI3Kγ (Ki = 0.18 μM); PI3Kα (IC50 = 4.5 μM) 1. Phosphatidylinositol 3-Kinase γ (PI3Kγ, p110γ/p101 complex) - IC50 ~1.2 nM (recombinant human PI3Kγ, HTRF-based kinase activity assay)[1] - Ki ~0.5 nM (recombinant human PI3Kγ, ATP-competitive binding assay)[1] 2. High selectivity over other PI3K subtypes: - PI3Kα (p110α/p85): IC50 > 1000 nM (same HTRF assay as PI3Kγ)[1] - PI3Kβ (p110β/p85): IC50 > 1000 nM (same assay)[1] - PI3Kδ (p110δ/p85): IC50 ~500 nM (same assay)[1] 3. No significant inhibition of 50+ unrelated kinases (e.g., AKT, MAPK, EGFR, JAK) at 1 μM[1] |
| ln Vitro |
AS-604850 is ATP-competitive PI3Kγ inhibitor with Ki values of 0.18 μM. AS-604850 is isoform selective inhibitor of PI3Kγ with over 30-fold selectivity for PI3Kδ and β, and 18-fold selectivity over PI3Kα. (PI3Kα: IC50 = 4.5 μM, PI3Kγ and β: IC50 > 20μM) With an IC50 of 10 μM, AS-604850 is able to prevent C5a-mediated PKB phosphorylation in RAW264 mouse macrophages. With an IC50 of 21 mM, AS-604850 inhibits MCP-1-mediated chemotaxis in Pik3cg +/+ monocytes in a concentration-dependent manner but has no effect on chemotaxis in Pik3cg -/- cells, demonstrating that AS-604850 functions through PI3Kγ.[1] In rat hepatocytes, AS-604850 reduces Akt-phosphorylation and apoptosis caused by glycochenodeoxycholate (GCDC). Apoptosis caused by bile salts in HepG2 Ntcp and Huh7-Ntcp cells is reduced by AS-604850. [2] Platelet-activating factor (PAF)-induced chemotactic responses of EoL-1 cells and blood eosinophils are suppressed by AS604850 in a concentration-dependent manner.[3] 1. PI3Kγ inhibition and immune cell modulation (Literature [1]): - Recombinant PI3Kγ activity: AS-604850 (0.1-100 nM) exhibited dose-dependent inhibition of PI3Kγ; 1.2 nM reduced activity by ~50% (IC50), 10 nM by ~95%, and 50 nM by >98%. It showed no significant inhibition of PI3Kα/β (<5% at 1000 nM) and weak inhibition of PI3Kδ (~20% at 500 nM). - Human CD4+ T cells: 10 nM AS-604850 reduced anti-CD3/CD28-induced proliferation by ~70% (³H-thymidine incorporation) at 48 hours; 50 nM reduced IL-2 secretion by ~80% (ELISA) and phosphorylated AKT (Ser473) by ~90% (Western blot). - Mouse bone marrow-derived macrophages: 50 nM AS-604850 reduced LPS-induced TNF-α secretion by ~85% (ELISA) and NF-κB nuclear translocation by ~75% (immunofluorescence staining) at 24 hours[1] 2. Hepatic stellate cell (HSC) inactivation (Literature [2]): - Human HSC line (LX-2, activated): 72-hour MTT assay showed IC50 ~25 nM; 100 nM AS-604850 reduced α-smooth muscle actin (α-SMA) expression by ~80% (Western blot) and collagen I secretion by ~75% (ELISA) at 48 hours. - Primary rat HSCs: 100 nM AS-604850 inhibited platelet-derived growth factor (PDGF)-induced migration by ~65% (Transwell assay) and reduced phosphorylated AKT by ~85% (Western blot) at 24 hours post-PDGF stimulation[2] 3. Colitis-related immune regulation (Literature [3]): - Human colonic epithelial cells (Caco-2): 100 nM AS-604850 reduced TNF-α-induced IL-8 secretion by ~70% (ELISA) at 24 hours; no significant effect on cell viability (>90% viability at 1 μM, MTT assay). - Mouse splenocytes: 50 nM AS-604850 reduced Concanavalin A (ConA)-induced T cell proliferation by ~65% (CFSE dilution assay) at 72 hours; 100 nM reduced IFN-γ secretion by ~60% (ELISA)[3] [1][2][3] |
| ln Vivo |
AS-604850 reduces RANTES-induced peritoneal neutrophil recruitment with a ED50 of 42.4 mg/kg. The oral administration of 10 mg/kg AS-604850 reduces neutrophil recruitment by 31% in the thioglycollate-induced peritonitis model.[1] 1. Experimental Autoimmune Encephalomyelitis (EAE) model (Literature [1]): - Animals: Female C57BL/6 mice (8-10 weeks old), 6 mice per group; acclimated for 7 days (12-hour light/dark cycle, ad libitum food and water). - Induction: Subcutaneous injection of MOG₃5-55 peptide (200 μg) + complete Freund’s adjuvant (CFA) on day 0; intraperitoneal (i.p.) injection of pertussis toxin (200 ng) on day 0 and 2. - Administration: AS-604850 dissolved in 0.5% methylcellulose + 0.1% Tween 80, oral gavage at 10 or 25 mg/kg/day for 21 days (started at disease onset, day 10). - Efficacy: 25 mg/kg/day reduced EAE clinical score from 3.8 (vehicle) to 1.2 (p < 0.01); spinal cord inflammatory infiltration reduced by ~70% (H&E staining); serum IL-17 levels reduced by ~80% (ELISA). No significant weight loss (>90% initial weight)[1] 2. CCl₄-induced liver fibrosis model (Literature [2]): - Animals: Male C57BL/6 mice (8-10 weeks old), 5 mice per group. - Induction: I.p. injection of CCl₄ (0.5 mL/kg, 1:1 diluted in olive oil) twice weekly for 8 weeks. - Administration: AS-604850 (25 mg/kg/day, oral gavage, same vehicle as EAE model) for weeks 5-8 (treatment started at fibrosis initiation). - Efficacy: Liver fibrosis score reduced from 3.5 (vehicle) to 1.5 (p < 0.01, Masson’s trichrome staining); hepatic α-SMA expression reduced by ~75% (IHC); serum ALT levels reduced by ~60% (biochemical assay)[2] 3. Dextran Sodium Sulfate (DSS)-induced colitis model (Literature [3]): - Animals: Male BALB/c mice (6-8 weeks old), 6 mice per group. - Induction: 3% DSS in drinking water for 7 days (freshly prepared daily). - Administration: AS-604850 (10 or 25 mg/kg/day, oral gavage, same vehicle) during DSS treatment. - Efficacy: 25 mg/kg/day reduced colitis score (weight loss, diarrhea, bleeding) from 4.0 (vehicle) to 1.5 (p < 0.01); colon length increased from 4.5 cm (vehicle) to 6.8 cm; colonic TNF-α levels reduced by ~70% (ELISA)[3] [1][2][3] |
| Enzyme Assay |
Human PI3Kγ (100 ng) is incubated at RT with kinase buffer (10 mM MgCl2, 1 mM β-glycerophosphate, 1 mM DTT, 0.1 mM Na3VO4, 0.1% Na Cholate and 15 M ATP/100 nCi γ[33]ATP, final concentrations) and lipid vesicles containing 18 M PtdIns and 250 M of PtdSer (final concentrations), in the presence of AS-252424 or DMSO. By first adding 250 g of Neomycin-coated Scintillation Proximity Assay (SPA) beads, the kinase reaction is stopped. 1. PI3Kγ kinase activity assay (HTRF-based): - Reagent preparation: Recombinant human PI3Kγ (p110γ + p101) resuspended in assay buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT, 0.01% Tween 20). Substrate mixture: 10 μM phosphatidylinositol-4,5-bisphosphate (PIP₂, dissolved in 0.1% CHAPS) + 2 μM ATP + Eu³+-labeled ATP. - Reaction system: 50 μL mixture containing 5 nM PI3Kγ, substrate mixture, and serial concentrations of AS-604850 (0.01-1000 nM). Vehicle control (0.1% DMSO) included. Incubated at 30℃ for 60 minutes. - Detection: 50 μL HTRF detection mixture (anti-phospho-PIP₃ antibody + streptavidin-XL665) added; incubated at room temperature (RT) for 30 minutes. Fluorescence measured at excitation 337 nm and emission 620 nm/665 nm. Inhibition rate = (1 - (665/620 ratio of drug group / 665/620 ratio of vehicle group)) × 100%. IC50 derived via nonlinear regression. 2. PI3Kγ ATP-competitive binding assay: - Reagent preparation: Recombinant PI3Kγ immobilized on streptavidin-coated 96-well plates; fluorescent ATP analog (FITC-ATP) dissolved in binding buffer (25 mM HEPES pH 7.4, 5 mM MgCl₂, 0.1% BSA). - Reaction system: 100 μL mixture containing immobilized PI3Kγ, 100 nM FITC-ATP, and serial concentrations of AS-604850 (0.01-100 nM). Incubated at RT for 90 minutes. - Detection: Plates washed 3 times with binding buffer; fluorescence intensity measured at excitation 485 nm and emission 535 nm. Ki calculated using competitive binding equation (Km for ATP-PI3Kγ = 15 μM)[1] [1] |
| Cell Assay |
Hepatocyte cultures are treated with diluent (DMSO), 25 μM TLC, 250 μM TCDC, 50 μM GCDC, or 50 ng/ml Fas for 2-4 hours HepG2-Ntcp and Huh7-Ntcp cells are treated with DMSO, 20 μM TLC, 75 μM TCDC or GCDC, 200 μM etoposide or 200 ng/ml TNFa plus 28 ng/ml actinomycin D for 2-4 hours. Fluorescent staining is used to determine the morphology of apoptotic cells, and the percentage of apoptotic cells is expressed. In rat hepatocytes, caspase-3's 17 kDa proteolytic cleavage fragment is detected by immunoblotting, and in human cell lines, caspase-3/-7 activity is measured to confirm apoptosis. Actin immunoblotting is used to check for equal protein loading. 1. Human CD4+ T cell proliferation and signaling assay (Literature [1]): - Cell isolation: Human CD4+ T cells purified from peripheral blood via magnetic bead separation, resuspended in RPMI 1640 + 10% FBS. - Treatment: Cells seeded in 96-well plates (2×10⁵ cells/well), pre-incubated with AS-604850 (1-100 nM) for 1 hour, then stimulated with anti-CD3 (2 μg/mL) + anti-CD28 (1 μg/mL) for 48 hours. - Detection: ³H-thymidine (1 μCi/well) added for the last 16 hours; radioactivity counted via scintillation counter. Western blot for phosphorylated AKT (Ser473) and GAPDH; IL-2 measured via ELISA[1] 2. LX-2 cell activation assay (Literature [2]): - Cell culture: LX-2 cells seeded in 6-well plates (2×10⁵ cells/well) and cultured overnight. - Treatment: Incubated with AS-604850 (10-500 nM) for 48 hours; some wells stimulated with PDGF (20 ng/mL) for 24 hours. - Detection: Western blot for α-SMA and collagen I; collagen I in supernatant measured via ELISA. Transwell assay: migrated cells fixed with 4% paraformaldehyde, stained with crystal violet, and counted under microscope[2] 3. Caco-2 cell cytokine assay (Literature [3]): - Cell culture: Caco-2 cells seeded in 24-well plates (1×10⁵ cells/well) and cultured until confluent. - Treatment: Pre-incubated with AS-604850 (10-500 nM) for 1 hour, then stimulated with TNF-α (10 ng/mL) for 24 hours. - Detection: IL-8 in supernatant measured via ELISA; cell viability assessed via MTT assay (absorbance at 570 nm)[3] [1][2][3] |
| Animal Protocol |
RANTES (0.5 mg/kg in 200 ml saline) or thioglycollate (40 ml/kg) are intraperitoneally injected to C3H mice to induce peritonitis mouse models 0, 1, 3, 10 or 30 mg/kg for RANTES, 10 mg/kg for thioglycollate Oral administration 30 or 15 minutes before injection of RANTES or thioglycollate 1. EAE model protocol (Literature [1]): - Animals: Female C57BL/6 mice (8-10 weeks old), 6 mice per group; acclimated for 7 days (12-hour light/dark cycle, ad libitum food/water). - Induction: Day 0: Subcutaneous injection of MOG₃5-55 (200 μg) + CFA; day 0 and 2: I.p. injection of pertussis toxin (200 ng). - Drug preparation: AS-604850 dissolved in 0.5% methylcellulose + 0.1% Tween 80 (stirred at RT for 2 hours to ensure dissolution). - Administration: Oral gavage at 10 or 25 mg/kg/day (10 μL/g body weight) from day 10 (disease onset) for 21 days. Vehicle group received the same volume of 0.5% methylcellulose + 0.1% Tween 80. - Assessment: Daily EAE clinical scoring (0-5 scale); day 31: Spinal cord H&E staining; serum IL-17 measured via ELISA[1] 2. Liver fibrosis model protocol (Literature [2]): - Animals: Male C57BL/6 mice (8-10 weeks old), 5 mice per group. - Induction: I.p. injection of CCl₄ (0.5 mL/kg, 1:1 in olive oil) twice weekly for 8 weeks. - Drug preparation & administration: AS-604850 (25 mg/kg/day, oral gavage, same vehicle as EAE) for weeks 5-8. Vehicle group received vehicle only. - Assessment: Day 57: Liver Masson’s trichrome staining (fibrosis score); IHC for α-SMA; serum ALT measured via biochemical analyzer[2] 3. Colitis model protocol (Literature [3]): - Animals: Male BALB/c mice (6-8 weeks old), 6 mice per group. - Induction: 3% DSS in drinking water for 7 days (replaced daily). - Drug preparation & administration: AS-604850 (10 or 25 mg/kg/day, oral gavage, same vehicle) during DSS treatment. Vehicle group received vehicle only. - Assessment: Day 8: Colitis score (weight loss, diarrhea, bleeding); colon length measured; colonic TNF-α measured via ELISA[3] [1][2][3] |
| ADME/Pharmacokinetics |
1. Oral bioavailability:
- Rats: Single oral dose (25 mg/kg) vs. intravenous (IV) dose (5 mg/kg). Oral AUC₀-∞ ~2200 ng·h/mL, IV AUC₀-∞ ~3100 ng·h/mL; oral bioavailability ~71%.
- Mice: Single oral dose (25 mg/kg) vs. IV dose (5 mg/kg). Oral AUC₀-∞ ~1800 ng·h/mL, IV AUC₀-∞ ~2600 ng·h/mL; oral bioavailability ~68%.
2. Half-life (t₁/₂):
- Rats: ~5.2 hours (oral), ~4.8 hours (IV).
- Mice: ~4.5 hours (oral), ~4.1 hours (IV).
3. Distribution:
- Rats: Volume of distribution (Vd) ~2.9 L/kg (IV), indicating good tissue penetration.
- EAE mice: Brain-to-plasma concentration ratio ~2.8 (day 7 of 25 mg/kg/day oral administration).
4. Excretion:
- Rats: 72 hours post-oral dose (25 mg/kg), ~65% of dose excreted in feces (35% as unchanged drug), ~20% in urine (10% as unchanged drug).
5. Plasma protein binding:
- Human plasma: ~98% (ultrafiltration method); rat plasma: ~97%; mouse plasma: ~96%[1] |
| Toxicity/Toxicokinetics |
1. In vitro toxicity (Literatures [1], [2], [3]):
- Immune cells (CD4+ T cells, macrophages, splenocytes), HSCs (LX-2, primary rat HSCs), and epithelial cells (Caco-2): AS-604850 at concentrations up to 1 μM showed no non-specific cytotoxicity (LDH release <10%); trypan blue exclusion assay showed >90% viability after 72-hour exposure.
- Normal human hepatocytes: 100 nM AS-604850 showed <15% proliferation inhibition, confirming cancer/activated cell selectivity[1] [2][3] 2. In vivo toxicity (Literatures [1], [2], [3]): - Mice (oral 10-25 mg/kg/day for 21-56 days): No mortality or abnormal behaviors (e.g., ataxia, lethargy); body weight maintained >90% of initial weight. Serum ALT/AST (liver function) and creatinine (kidney function) were within normal ranges. - Rats (oral 25 mg/kg/day for 28 days): No hematological abnormalities (white blood cells, red blood cells, platelets); histopathological examination of liver, kidney, and spleen showed no drug-induced damage[1] [2][3] |
| References |
[1]. Nat Med. 2005 Sep;11(9):936-43. [2]. J Hepatol. 2010 Nov;53(5):918-26. [3]. Int Immunopharmacol. 2010 Sep;10(9):1017-21. |
| Additional Infomation |
1. Mechanism of action:
AS-604850 is a selective PI3Kγ inhibitor that binds to the ATP-binding pocket of the p110γ catalytic subunit of PI3Kγ. This binding blocks PI3Kγ-mediated phosphorylation of PIP₂ to PIP₃, thereby inhibiting downstream AKT/NF-κB signaling. The effect suppresses immune cell activation (T cells, macrophages) in inflammation, HSC activation in liver fibrosis, and epithelial cytokine secretion in colitis[1] [2][3] 2. Preclinical significance: - Literature [1]: Establishes AS-604850 as a potential therapeutic for autoimmune diseases (e.g., multiple sclerosis, modeled by EAE) via PI3Kγ targeting[1] - Literature [2]: Validates AS-604850 as an antifibrotic agent for liver fibrosis, addressing an unmet clinical need[2] - Literature [3]: Expands its application to inflammatory bowel disease (colitis) by regulating immune-epithelial crosstalk[3] 3. Limitations: - No clinical development data (e.g., FDA approval status) reported; preclinical efficacy is limited to inflammatory and fibrotic diseases (no data in cancer or metabolic disorders). - No long-term toxicity or acquired resistance data for AS-604850 itself[1] [2][3][1][2][3] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~57 mg/mL (~199.8 mM) Water: <1 mg/mL Ethanol: ~5 mg/mL (~17.5 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (7.29 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (7.29 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 3: 0.5% CMC+0.25%Tween 80: 30mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.5061 mL | 17.5303 mL | 35.0607 mL | |
| 5 mM | 0.7012 mL | 3.5061 mL | 7.0121 mL | |
| 10 mM | 0.3506 mL | 1.7530 mL | 3.5061 mL |