Physicochemical Properties
| Molecular Formula | C16H18N2O |
| Molecular Weight | 254.33 |
| Exact Mass | 254.142 |
| CAS # | 71308-34-4 |
| PubChem CID | 9943022 |
| Appearance | Light yellow to yellow solid powder |
| LogP | 4.069 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 3 |
| Rotatable Bond Count | 3 |
| Heavy Atom Count | 19 |
| Complexity | 396 |
| Defined Atom Stereocenter Count | 0 |
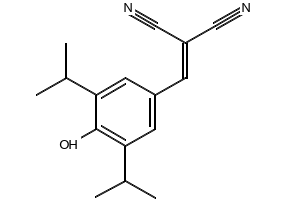
| SMILES | OC1=C(C(C)C)C=C(/C=C(\C#N)/C#N)C=C1C(C)C |
| InChi Key | JSDJFOADLLJFQX-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C16H18N2O/c1-10(2)14-6-12(5-13(8-17)9-18)7-15(11(3)4)16(14)19/h5-7,10-11,19H,1-4H3 |
| Chemical Name | 2-(4-hydroxy-3,5-diisopropylbenzylidene)malononitrile |
| Synonyms | AG-1406AG 1406AG1406CTK2G2659CTK2G-2659CTK2G 2659 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Epidermal Growth Factor Receptor (EGFR) tyrosine kinase (IC50 = 0.04 μM); Platelet-Derived Growth Factor Receptor (PDGFR) tyrosine kinase (IC50 > 10 μM); Vascular Endothelial Growth Factor Receptor (VEGFR) tyrosine kinase (IC50 > 10 μM) [1] |
| ln Vitro |
AG 1406 potently inhibited recombinant EGFR tyrosine kinase activity in a dose-dependent manner, with IC50 = 0.04 μM; it showed no significant inhibition of PDGFR or VEGFR tyrosine kinase (IC50 > 10 μM) [1] - In EGFR-overexpressing tumor cell lines: AG 1406 inhibited proliferation of A431 (epidermoid carcinoma) with IC50 = 0.1 μM, MDA-MB-468 (breast cancer) with IC50 = 0.2 μM, and KB (oral carcinoma) with IC50 = 0.15 μM (MTT assay) [1] - Western blot analysis showed that AG 1406 (0.1-1 μM, 6 h) dose-dependently suppressed EGF-induced EGFR phosphorylation (Tyr1173) in A431 cells: 1 μM reduced phosphorylation by ~92% compared to EGF-stimulated control; downstream signaling molecules (p-Akt, p-ERK1/2) were also inhibited by ~85% and ~80% respectively [1] - AG 1406 (0.5 μM, 72 h) induced G1 cell cycle arrest in A431 cells (flow cytometry): G1 phase cells increased from 42% (control) to 68%, S phase cells decreased from 35% to 17% [1] - In colony formation assay, AG 1406 (0.05-0.2 μM) dose-dependently reduced the number of A431 cell colonies: 0.2 μM inhibited colony formation by ~75% compared to control [1] |
| ln Vivo |
In nude mice bearing A431 xenografts: Oral administration of AG 1406 (50 mg/kg, once daily) for 21 days significantly inhibited tumor growth; tumor volume at day 21 was 32% of vehicle control (P<0.01), and tumor weight was reduced by ~68% compared to vehicle [1] - In MDA-MB-468 breast cancer xenograft mice: AG 1406 (75 mg/kg, oral, daily for 18 days) suppressed tumor growth by ~62% (tumor volume) and ~60% (tumor weight) versus vehicle (P<0.01) [1] - Immunohistochemical staining of A431 tumor tissues from AG 1406-treated mice showed reduced EGFR phosphorylation (by ~70%) and Ki-67 (cell proliferation marker) expression (by ~65%) compared to vehicle [1] |
| Enzyme Assay |
Recombinant human EGFR tyrosine kinase domain was purified and resuspended in kinase buffer. AG 1406 was serially diluted (0.001-10 μM) and mixed with the recombinant kinase. The reaction mixture was supplemented with MgCl2, [γ-32P]ATP (at Km concentration for EGFR), and a synthetic peptide substrate (derived from EGFR autophosphorylation site). After incubation at 30°C for 30 minutes, the reaction was terminated by spotting onto phosphocellulose filters. Filters were washed to remove unincorporated radioactivity, and bound radioactivity (indicating phosphorylated substrate) was measured by scintillation counting. IC50 values were calculated by nonlinear regression of dose-response inhibition curves [1] - For PDGFR and VEGFR kinase assays: The same protocol was followed using recombinant PDGFR/VEGFR tyrosine kinase domains and their respective peptide substrates, with AG 1406 tested at concentrations up to 10 μM [1] |
| Cell Assay |
Cell proliferation assay: Tumor cells (A431, MDA-MB-468, KB) were seeded in 96-well plates (5×103 cells/well) and cultured overnight. AG 1406 (serial dilutions: 0.01-10 μM) was added, and cells were incubated for 72 h. MTT reagent was added, incubated for 4 h, formazan crystals were dissolved in DMSO, and absorbance was measured at 570 nm. IC50 values were derived from dose-response curves [1] - Western blot assay for signaling pathways: A431 cells were seeded in 6-well plates (2×105 cells/well) and serum-starved overnight. Cells were pre-treated with AG 1406 (0.1-1 μM) for 1 h, then stimulated with EGF (100 ng/mL) for 15 minutes. Cells were lysed, proteins were separated by SDS-PAGE, transferred to PVDF membranes, and probed with antibodies against p-EGFR (Tyr1173), EGFR, p-Akt (Ser473), Akt, p-ERK1/2, and ERK1/2. Band intensity was quantified by densitometry [1] - Cell cycle analysis: A431 cells were treated with AG 1406 (0.5 μM) for 72 h, fixed with ethanol, stained with propidium iodide, and analyzed by flow cytometry to determine cell cycle distribution [1] - Colony formation assay: A431 cells were seeded in 6-well plates (200 cells/well) and allowed to attach overnight. AG 1406 (0.05-0.2 μM) was added, and cells were cultured for 14 days. Colonies were fixed with formaldehyde, stained with crystal violet, and counted manually [1] |
| Animal Protocol |
Xenograft model establishment: Female nude mice (6-8 weeks old) were subcutaneously injected with 5×106 A431 or MDA-MB-468 cells (suspended in PBS/matrigel) into the right flank. Tumors were allowed to grow to ~100 mm3 before initiating treatment [1] - Drug administration: Mice were randomly divided into vehicle control and AG 1406 treatment groups (n=8/group). AG 1406 was dissolved in 10% DMSO + 90% polyethylene glycol 400 (PEG400) and administered orally via gavage. A431-bearing mice received 50 mg/kg once daily for 21 days; MDA-MB-468-bearing mice received 75 mg/kg once daily for 18 days. Vehicle control mice received the same volume of 10% DMSO + 90% PEG400 [1] - Tumor and body weight monitoring: Tumor volume was measured every 3 days using calipers (volume = length × width² / 2). Body weight was recorded weekly to assess general toxicity [1] - Tissue collection: At the end of treatment, mice were euthanized, tumors were excised, weighed, and fixed in formalin for immunohistochemical analysis [1] |
| Toxicity/Toxicokinetics |
In nude mice treated with AG 1406 (up to 75 mg/kg, oral, daily for 21 days), no significant weight loss (≤5% of initial weight) or gross abnormalities in major organs (liver, kidney, spleen) were observed [1] - Plasma protein binding rate of AG 1406 in mice was 91% ± 3% (ultrafiltration method) [1] |
| References |
[1]. Methods and compositions for inhibiting cell proliferative disorders. Patent, US5789427A. |
| Additional Infomation |
AG 1406 is a synthetic small-molecule inhibitor belonging to the quinazoline class, which binds competitively to the ATP-binding pocket of EGFR tyrosine kinase [1] - The chemical structure of AG 1406 features a quinazoline core with a 4-anilino substituent, which is critical for high affinity and selectivity for EGFR [1] - AG 1406 exerts antitumor effects by inhibiting EGFR-mediated signaling pathways, thereby blocking tumor cell proliferation, inducing cell cycle arrest, and suppressing tumor angiogenesis [1] - The patent discloses AG 1406 as a potential therapeutic agent for the treatment of EGFR-overexpressing solid tumors, including lung cancer, breast cancer, head and neck cancer, and colorectal cancer [1] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.9319 mL | 19.6595 mL | 39.3190 mL | |
| 5 mM | 0.7864 mL | 3.9319 mL | 7.8638 mL | |
| 10 mM | 0.3932 mL | 1.9659 mL | 3.9319 mL |