AC710 is a potent, orally bioactive, and selective inhibitor of PDGFR (platelet-derived growth factor receptor) family kinase with Kd values of 0.6, 1.57, 1, 1.3, 1.0 nM for FLT3, CSF1R, KIT, PDGFRα and PDGFRβ, respectively. As a PDGFR inhibitor, AC710 has potential anticancer activity.Because it inhibits PDGFR, AC710 may have anticancer properties. The preclinical development candidate for AC710 was chosen based on the outcomes of a seven-day in vivo tolerability study, a collagen-induced arthritis model, and a mouse tumor xenograft.
Physicochemical Properties
| Molecular Formula | C31H42N6O4 | |
| Molecular Weight | 562.7 | |
| Exact Mass | 562.326 | |
| Elemental Analysis | C, 66.17; H, 7.52; N, 14.94; O, 11.37 | |
| CAS # | 1351522-04-7 | |
| Related CAS # | AC710 Mesylate;1351522-05-8 | |
| PubChem CID | 54760053 | |
| Appearance | White to off-white solid powder | |
| Density | 1.2±0.1 g/cm3 | |
| Boiling Point | 580.4±50.0 °C at 760 mmHg | |
| Flash Point | 304.8±30.1 °C | |
| Vapour Pressure | 0.0±1.6 mmHg at 25°C | |
| Index of Refraction | 1.591 | |
| LogP | 4.86 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 7 | |
| Rotatable Bond Count | 8 | |
| Heavy Atom Count | 41 | |
| Complexity | 880 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | O(C1=C([H])N=C(C(N([H])C2C([H])=C([H])C(=C([H])C=2[H])N([H])C(N([H])C2C([H])=C(C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H])ON=2)=O)=O)C([H])=C1[H])C1([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])N(C([H])([H])C([H])([H])[H])C(C([H])([H])[H])(C([H])([H])[H])C1([H])[H] |
|
| InChi Key | JVCWPUFNLFSKFS-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C31H42N6O4/c1-9-37-30(5,6)17-23(18-31(37,7)8)40-22-14-15-24(32-19-22)27(38)33-20-10-12-21(13-11-20)34-28(39)35-26-16-25(41-36-26)29(2,3)4/h10-16,19,23H,9,17-18H2,1-8H3,(H,33,38)(H2,34,35,36,39) | |
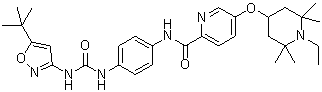
| Chemical Name | N-[4-[(5-tert-butyl-1,2-oxazol-3-yl)carbamoylamino]phenyl]-5-(1-ethyl-2,2,6,6-tetramethylpiperidin-4-yl)oxypyridine-2-carboxamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PDGFRα (Kd = 1.3 nM); PDGFRβ (Kd = 1 nM); c-Kit (Kd = 1 nM); FLT3 (Kd = 0.6 nM); CSF1R (Kd = 1.57 nM)
AC710 targets platelet-derived growth factor receptor (PDGFR) family kinases, including PDGFRα (IC50 = 1.2 nM), PDGFRβ (IC50 = 0.8 nM), and colony-stimulating factor 1 receptor (CSF-1R, IC50 = 2.5 nM) [1] AC710 exhibits >100-fold selectivity over non-PDGFR family kinases, including VEGFR2 (IC50 = 156 nM), EGFR (IC50 = 218 nM), FGFR1 (IC50 = 189 nM), and c-Met (IC50 = 245 nM) [1] |
| ln Vitro |
AC710 is a potent, orally active, and selective inhibits the PDGFR (platelet-derived growth factor receptor) family kinase, with Kd values of 0.6, 1.57, 1, 1.3, and 1.0 nM for FLT3, CSF1R, KIT, PDGFRα, and PDGFRβ, individually. AC710 may have anticancer properties because it is a PDGFR inhibitor. The preclinical development candidate AC710 was chosen based on outcomes from a rat in vivo tolerability study, a mouse tumor xenograft, and a collagen-induced arthritis model. In recombinant kinase activity assays, AC710 dose-dependently inhibited PDGFRα, PDGFRβ, and CSF-1R with IC50 values of 1.2 nM, 0.8 nM, and 2.5 nM, respectively, by competing with ATP for binding to the kinase domain [1] - In Ba/F3 cells engineered to express human PDGFRα (Ba/F3-PDGFRα) or PDGFRβ (Ba/F3-PDGFRβ), AC710 inhibited cell proliferation with IC50 values of 3.5 nM and 2.8 nM, respectively, after 72-hour treatment (CCK-8 assay) [1] - In human glioblastoma U87-MG cells (expressing endogenous PDGFRβ) and colon cancer HCT116 cells (expressing PDGFRα), AC710 suppressed proliferation with IC50 values of 4.2 nM and 5.7 nM, respectively [1] - Western blot analysis showed that AC710 (5 nM) inhibited PDGFRα/β phosphorylation (p-PDGFRα/β) by ~85% and ~90% in Ba/F3-PDGFRα and Ba/F3-PDGFRβ cells, respectively; downstream AKT and ERK1/2 phosphorylation was reduced by ~70-75% and ~65-70%, respectively [1] - AC710 (up to 100 nM) had no significant effect on the viability of normal human dermal fibroblasts (NHDF) or human umbilical vein endothelial cells (HUVECs), indicating low toxicity to normal cells [1] |
| ln Vivo |
AC710 shows a dose-dependent effect on disease in a mouse model of collagen-induced arthritis (CIA), starting at a dose of 3 mg/kg and lasting for 15 days (days 0–14). When given at a safe dosage, dexomethasone reduces joint swelling and inflammation more effectively than AC710, either equally or marginally better. At the tested doses, AC710 is well tolerated[1]. Tumor growth is momentarily stopped at 0.3 mg/kg of AC710, but it soon resumes. Tumors fully regress at 3 and 30 mg/kg of AC710, and the tumor volume remains suppressed long after dosing is stopped. Animals treated with AC710 at all doses show no signs of body weight loss, suggesting that mice can tolerate it well at effective dosages. In nude mice bearing Ba/F3-PDGFRβ xenografts, oral administration of AC710 (10 mg/kg/day, 20 mg/kg/day, or 40 mg/kg/day for 14 days) dose-dependently inhibited tumor growth: high-dose treatment resulted in a tumor growth inhibition (TGI) rate of 78% and reduced tumor weight from 0.92 ± 0.15 g (vehicle) to 0.20 ± 0.04 g [1] - In U87-MG human glioblastoma xenograft mice, oral AC710 (20 mg/kg/day for 21 days) achieved a TGI rate of 65% and decreased tumor microvessel density (MVD) by ~55% (immunohistochemical staining for CD31) compared to vehicle control [1] - No significant changes in body weight (vehicle: 23.1 ± 1.2 g vs. high-dose: 22.5 ± 1.0 g) or histopathological abnormalities in major organs (liver, kidney, heart, lung) were observed in treated mice [1] |
| Enzyme Assay |
AC710 is a potent, orally active, and selective inhibits the PDGFR (platelet-derived growth factor receptor) family kinase, with Kd values of 0.6, 1.57, 1, 1.3, and 1.0 nM for FLT3, CSF1R, KIT, PDGFRα, and PDGFRβ, individually. PDGFR family kinase activity assay: Recombinant human PDGFRα, PDGFRβ, and CSF-1R kinase domains were individually incubated with reaction buffer containing ATP (10 μM) and a fluorescent-labeled peptide substrate. Serial dilutions of AC710 (0.001-1000 nM) were added to the reaction mixture, which was incubated at 37°C for 45 minutes. The reaction was terminated by adding a stop solution containing EDTA, and fluorescence intensity (excitation 485 nm, emission 535 nm) was measured to assess kinase-mediated peptide phosphorylation. IC50 values were calculated by nonlinear regression of dose-response curves [1] - Kinase selectivity assay: A panel of 30 recombinant non-PDGFR family kinases (including VEGFR2, EGFR, FGFR1, c-Met) was subjected to the same kinase assay protocol. AC710 (0.001-1000 nM) was tested to determine IC50 values for these kinases, confirming the selective inhibition of PDGFR family members [1] |
| Cell Assay |
Ba/F3 cell proliferation assay: Ba/F3-PDGFRα and Ba/F3-PDGFRβ cells were seeded in 96-well plates at 3×10³ cells/well. After 24 hours of culture, serial dilutions of AC710 (0.1-100 nM) were added, and cells were incubated for another 72 hours. CCK-8 reagent was added, and absorbance at 450 nm was measured to calculate cell viability and IC50 values [1] - Tumor cell proliferation assay: U87-MG and HCT116 cells were seeded in 96-well plates at 5×10³ cells/well. Following 24-hour attachment, AC710 (0.1-100 nM) was added, and cells were cultured for 72 hours. Cell viability was assessed by CCK-8 assay, and IC50 values were determined [1] - Western blot assay: Ba/F3-PDGFRα and Ba/F3-PDGFRβ cells were treated with AC710 (0.5-50 nM) for 24 hours. Cells were lysed in RIPA buffer, and proteins were separated by SDS-PAGE. Membranes were probed with antibodies against p-PDGFRα, PDGFRα, p-PDGFRβ, PDGFRβ, p-AKT, AKT, p-ERK1/2, ERK1/2, and GAPDH (loading control). Chemiluminescent detection and densitometric analysis were used to quantify protein expression levels [1] - Normal cell viability assay: NHDF and HUVECs were seeded in 96-well plates at 5×10³ cells/well, treated with AC710 (0.1-100 nM) for 72 hours, and cell viability was measured by CCK-8 assay [1] |
| Animal Protocol |
Mice: In athymic nude mice, the antitumor efficacy of AC710 is evaluated using the MV4-11cell line in a subcutaneous flank-tumor xenograft model. Over the course of two weeks, AC710 is dosed at0.3,3, and 30 mg/kg. Both body weight and tumor growth are tracked. Ba/F3-PDGFRβ xenograft model: Female BALB/c nude mice (4-6 weeks old) were subcutaneously implanted with 5×10⁶ Ba/F3-PDGFRβ cells. When tumors reached a volume of ~100 mm³, mice were randomly divided into vehicle control, AC710 10 mg/kg, 20 mg/kg, and 40 mg/kg groups (n=6 per group). The drug was dissolved in 0.5% methylcellulose + 0.2% Tween 80 and administered by oral gavage once daily for 14 days. Tumor volume was measured every 2 days using calipers, and tumor weight was recorded at the end of treatment [1] - U87-MG xenograft model: Female nude mice (4-6 weeks old) were subcutaneously implanted with 1×10⁷ U87-MG cells. When tumors reached ~150 mm³, mice were assigned to vehicle or AC710 20 mg/kg groups (n=7 per group). The drug was formulated as described above and administered orally once daily for 21 days. Tumor volume was monitored twice weekly, and tumor tissues were collected for CD31 immunohistochemical staining [1] |
| ADME/Pharmacokinetics |
Oral bioavailability: In mice, oral administration of AC710 (20 mg/kg) resulted in an oral bioavailability of ~62% [1] - Plasma half-life (t1/2): In mice, the terminal plasma half-life of AC710 was 4.5 ± 0.6 hours after oral administration (20 mg/kg) [1] - Peak plasma concentration (Cmax): In mice, oral AC710 (20 mg/kg) achieved a Cmax of 387 ± 42 ng/mL at 1.0 ± 0.2 hours post-dosing [1] - Area under the plasma concentration-time curve (AUC0-∞): In mice, AUC0-∞ of AC710 was 1890 ± 210 ng·h/mL after a single oral dose of 20 mg/kg [1] - Volume of distribution (Vd/F): In mice, the apparent volume of distribution was 12.8 ± 1.5 L/kg (oral 20 mg/kg) [1] - Clearance (CL/F): Apparent oral clearance in mice was 10.6 ± 1.2 mL/min/kg (oral 20 mg/kg) [1] |
| Toxicity/Toxicokinetics |
In vitro cytotoxicity: AC710 exhibited CC50 values >100 nM in NHDF and HUVECs, indicating low toxicity to normal cells [1] - Acute toxicity in mice: Single oral administration of AC710 up to 200 mg/kg did not cause mortality or overt toxicity (lethargy, weight loss, abnormal behavior) [1] - Chronic toxicity in mice: Repeated oral administration of AC710 (40 mg/kg/day for 14 days) did not induce significant changes in hematological parameters (red blood cells, white blood cells, platelets) or serum biochemical markers (ALT, AST, creatinine, BUN) [1] - Plasma protein binding: AC710 exhibited plasma protein binding of 93-95% in mouse plasma and 94-96% in human plasma (equilibrium dialysis) [1] |
| References |
[1]. Discovery of AC710, a Globally Selective Inhibitor of Platelet-Derived Growth Factor Receptor-Family Kinases. ACS Med Chem Lett. 2012 Sep 24;3(12):997-1002. |
| Additional Infomation |
AC710 is a potent, orally active, and globally selective small-molecule inhibitor of PDGFR family kinases (PDGFRα, PDGFRβ, CSF-1R) [1] - The therapeutic mechanism of AC710 involves selective inhibition of PDGFR family kinase activity, blocking downstream PI3K-AKT and Ras-ERK signaling pathways, thereby suppressing proliferation and survival of PDGFR-driven tumor cells and inhibiting tumor angiogenesis [1] - AC710 was developed for the treatment of solid tumors driven by aberrant PDGFR family kinase activation, including glioblastoma, colon cancer, and other PDGFR-dependent malignancies [1] - Preclinical data demonstrate that AC710 exhibits strong in vitro and in vivo antitumor activity with high selectivity for PDGFR family kinases and favorable safety profiles, supporting its potential as a targeted cancer therapy [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 0.5 mg/mL (0.89 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 5.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 0.5 mg/mL (0.89 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 5.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 0.5 mg/mL (0.89 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 5.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7771 mL | 8.8857 mL | 17.7715 mL | |
| 5 mM | 0.3554 mL | 1.7771 mL | 3.5543 mL | |
| 10 mM | 0.1777 mL | 0.8886 mL | 1.7771 mL |