Physicochemical Properties
| Molecular Formula | C30H44N4O10S |
| Molecular Weight | 652.76 |
| Exact Mass | 652.277 |
| Elemental Analysis | C, 55.20; H, 6.79; N, 8.58; O, 24.51; S, 4.91 |
| CAS # | 1196800-40-4 |
| Related CAS # | Omadacycline;389139-89-3;Omadacycline tosylate;1075240-43-5;Omadacycline hydrochloride;1196800-39-1;Omadacycline-d9;2272886-41-4 |
| PubChem CID | 74891320 |
| Appearance | Solid powder |
| Hydrogen Bond Donor Count | 7 |
| Hydrogen Bond Acceptor Count | 13 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 45 |
| Complexity | 1230 |
| Defined Atom Stereocenter Count | 4 |
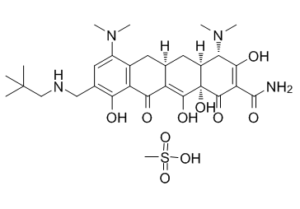
| SMILES | CS(=O)(O)=O.O=C(C(C1=O)=C(O)[C@@H](N(C)C)[C@]2([H])C[C@]3([H])CC4=C(C(C3=C(O)[C@@]21O)=O)C(O)=C(CNCC(C)(C)C)C=C4N(C)C)N |
| InChi Key | BRTZQVQPPVIFKG-XGLFQKEBSA-N |
| InChi Code | InChI=1S/C29H40N4O7.CH4O3S/c1-28(2,3)12-31-11-14-10-17(32(4)5)15-8-13-9-16-21(33(6)7)24(36)20(27(30)39)26(38)29(16,40)25(37)18(13)23(35)19(15)22(14)34;1-5(2,3)4/h10,13,16,21,31,34-35,38,40H,8-9,11-12H2,1-7H3,(H2,30,39);1H3,(H,2,3,4)/t13-,16-,21-,29-;/m0./s1 |
| Chemical Name | (4S,4aS,5aR,12aR)-4,7-Bis(dimethylamino)-9-[(2,2-dimethylpropylamino)methyl]-1,10,11,12a-tetrahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4H-tetracene-2-carboxamide; mesylate |
| Synonyms | PTK-0796; PTK 0796; PTK0796; (4S,4aS,5aR,12aR)-4,7-bis(dimethylamino)-9-[(2,2-dimethylpropylamino)methyl]-1,10,11,12a-tetrahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4H-tetracene-2-carboxamide;methanesulfonic acid; PTK 0796 mesylate; Nuzyra; Amadacyclin; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture and light. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Tetracycline; protein synthesis of bacteria | ||
| ln Vitro | Omadacycline is a novel, aminomethyl tetracycline antibiotic being developed for oral and intravenous (IV) administration to treat community-acquired bacterial infections such as acute bacterial skin and skin structure infections (ABSSSI), community-acquired bacterial pneumonia (CABP), and urinary tract infections (UTI). In vitro, omadacycline has activity against Gram-positive and Gram-negative aerobes, anaerobes, and atypical pathogens including Legionella and Chlamydia spp. Omadacycline offers once daily oral and IV dosing and a clinical tolerability and safety profile that compares favorably with contemporary antibiotics used across serious community-acquired infections where resistance has rendered many less effective. In studies in patients with complicated skin and skin structure infections, including those with MRSA infections, omadacycline exhibited an efficacy and tolerability profile that was comparable to linezolid. Ongoing and planned clinical studies are evaluating omadacycline as monotherapy for treating serious community-acquired bacterial infections including Acute Bacterial Skin and Skin Structure Infections (ABSSSI) and Community-Acquired Bacterial Pneumonia (CABP). This review provides an overview of the discovery, microbiology, nonclinical data, and available clinical safety and efficacy data for omadacycline, with reference to other contemporary tetracycline-derived antibiotics. | ||
| ln Vivo | In vivo efficacy of omadacycline is demonstrated using an intraperitoneal infection model in mice. A single intravenous dose of omadacycline exhibits efficacy against Streptococcus pneumoniae, Escherichia coli, and Staphylococcus aureus, including tet (M) and tet (K) efflux-containing strains and MRSA strains. The 50% effective doses (ED50s) for Streptococcus pneumoniae obtained ranged from 0.45 mg/kg to 3.39 mg/kg, the ED50s for Staphylococcus aureus obtained ranges from 0.30 mg/kg to 1.74 mg/kg, and the ED50 for Escherichia coli is 2.02 mg/kg. | ||
| Enzyme Assay |
In vitro stability and drug–drug interaction potential of omadacycline[5] The stability of omadacycline (4.8 and 48 μM) was assessed in human microsomes and hepatocytes. After 30 min incubation of omadacycline in human microsomes, >90% of omadacycline was recovered intact. Similarly, after incubation of omadacycline up to 24 h in human hepatocytes, >86% was recovered intact. These results indicate that omadacycline is not metabolized to any significant extent. The potential for drug-drug-interactions with omadacycline was assessed using either pooled human liver microsome preparations, S9, liver cytosol, or recombinant flavin monooxygenases (FMO1, FMO3, FMO5). Induction of CYP450 isozymes was evaluated in primary human hepatocytes incubated with omadacycline 1–100 μM and a substrate probe for 24 and 48 h. Inhibition of CYP450 isozymes was evaluated with pooled human microsomes at omadacycline concentrations of 1–50 μM and isozyme specific substrates at concentrations approximating the Km of each substrate. Isozymes evaluated included CYP 1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 2J2, and 3A4/5. Omadacycline did not induce CYP isozymes, and no or minimal (<40% of maximal positive control response) induction of their mRNAs was observed. Omadacycline demonstrated no significant inhibition of CYP isozyme activity. In addition, there was no time-dependent inhibition of omadacycline or its possible metabolites for CYP1A2 2C9, 2D6 or 3A4/5.[5] |
||
| Cell Assay | The omadacycline MIC90s for MRSA, VRE, and beta-hemolytic streptococci are 1.0 μg/mL, 0.25 μg/mL, and 0.5 μg/mL, respectively, and the omadacycline MIC90s for PRSP and H. influenzae are 0.25 μg/ml and 2.0 μg/mL, respectively. Omadacycline is active against organisms demonstrating the two major mechanisms of resistance, ribosomal protection and active tetracycline efflux[1]. Omadacycline inhibits protein synthesis while having no significant effect on RNA, DNA and peptidoglycan synthesis. Further, omadacycline binds to the tetracycline binding site on the 30S subunit of the bacterial ribosome with enhanced binding similar to tigecycline based on additional molecular interactions. | ||
| Animal Protocol |
|
||
| ADME/Pharmacokinetics | The pharmacokinetics of omadacycline are best described by a linear, three-compartment model following a zero-order intravenous infusion or first-order oral administration with transit compartments to account for delayed absorption. Omadacycline has a volume of distribution (Vd) ranging from 190 to 204 L, a terminal elimination half-life (t½) of 13.5-17.1 h, total clearance (CLT) of 8.8-10.6 L/h, and protein binding of 21.3% in healthy subjects. Oral bioavailability of omadacycline is estimated to be 34.5%. A single oral dose of 300 mg (bioequivalent to 100 mg IV) of omadacycline administered to fasted subjects achieved a maximum plasma concentration (Cmax) of 0.5-0.6 mg/L and an area under the plasma concentration-time curve from 0 to infinity (AUC0-∞) of 9.6-11.9 mg h/L. The free plasma area under concentration-time curve divided by the minimum inhibitory concentration (i.e., fAUC24h/MIC), has been established as the pharmacodynamic parameter predictive of omadacycline antibacterial efficacy. Several animal models including neutropenic murine lung infection, thigh infection, and intraperitoneal challenge model have documented the in vivo antibacterial efficacy of omadacycline. A phase II clinical trial on complicated skin and skin structure infection (cSSSI) and three phase III clinical trials on ABSSSI and CABP demonstrated the safety and efficacy of omadacycline. The phase III trials, OASIS-1 (ABSSSI), OASIS-2 (ABSSSI), and OPTIC (CABP), established non-inferiority of omadacycline to linezolid (OASIS-1, OASIS-2) and moxifloxacin (OPTIC), respectively. Omadacycline is currently approved by the FDA for use in treatment of ABSSSI and CABP. Phase II clinical trials involving patients with acute cystitis and acute pyelonephritis are in progress. Mild, transient gastrointestinal events are the predominant adverse effects associated with use of omadacycline. Based on clinical trial data to date, the adverse effect profile of omadacycline is similar to studied comparators, linezolid and moxifloxacin. Unlike tigecycline and eravacycline, omadacycline has an oral formulation that allows for step-down therapy from the intravenous formulation, potentially facilitating earlier hospital discharge, outpatient therapy, and cost savings. Omadacycline has a potential role as part of an antimicrobial stewardship program in the treatment of patients with infections caused by antibiotic-resistant and multidrug-resistant Gram-positive [including methicillin-resistant Staphylococcus aureus (MRSA)] and Gram-negative pathogens. [https://pubmed.ncbi.nlm.nih.gov/31970713/] | ||
| Toxicity/Toxicokinetics |
Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No information is available on the use of omadacycline during breastfeeding. It is unknown how much omadacycline is excreted into breastmilk, but the drug is only about 35% absorbed orally under optimal circumstances, and is probably less from milk because of its calcium content. The manufacturer states that breastfeeding is not recommended during treatment and for 4 days after the last dose. If an infant is breastfed, monitor the infant for possible effects on the gastrointestinal flora, such as diarrhea, candidiasis (e.g., thrush, diaper rash) or rarely, blood in the stool indicating possible antibiotic-associated colitis. As a theoretical precaution, avoid prolonged or repeat courses during nursing. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. |
||
| References |
[1]. Pharmaceuticals (Basel). 2019 Apr 21;12(2):63. [2]. Antimicrob Agents Chemother. 2014;58(2):1127-35. [3]. Drugs. 2020 Feb;80(3):285-313. [4]. Drugs. 2018 Dec;78(18):1931-1937. [5]. Bioorg Med Chem.2016 Dec 15;24(24):6409-6419. |
||
| Additional Infomation |
Omadacycline (Nuzyra®) is a new aminomethylcycline, approved by the U. S. Food and Drug Administration in 2018, as a tetracycline antibacterial. It can be used in community-acquired pneumonia and in acute bacterial skin and skin-structure infections. It was developed and is commercialized by Paratek Pharmaceuticals. It is a semisynthetic compound, derived from minocycline, capable of evading widely distributed efflux and target protection antibacterial resistance mechanisms and has demonstrated activity in a broad spectrum of bacteria.[1] Omadacycline is the first intravenous and oral 9-aminomethylcycline in clinical development for use against multiple infectious diseases including acute bacterial skin and skin structure infections (ABSSSI), community-acquired bacterial pneumonia (CABP), and urinary tract infections (UTI). The comparative in vitro activity of omadacycline was determined against a broad panel of Gram-positive clinical isolates, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus (VRE), Lancefield groups A and B beta-hemolytic streptococci, penicillin-resistant Streptococcus pneumoniae (PRSP), and Haemophilus influenzae (H. influenzae). The omadacycline MIC90s for MRSA, VRE, and beta-hemolytic streptococci were 1.0 μg/ml, 0.25 μg/ml, and 0.5 μg/ml, respectively, and the omadacycline MIC90s for PRSP and H. influenzae were 0.25 μg/ml and 2.0 μg/ml, respectively. Omadacycline was active against organisms demonstrating the two major mechanisms of resistance, ribosomal protection and active tetracycline efflux. In vivo efficacy of omadacycline was demonstrated using an intraperitoneal infection model in mice. A single intravenous dose of omadacycline exhibited efficacy against Streptococcus pneumoniae, Escherichia coli, and Staphylococcus aureus, including tet(M) and tet(K) efflux-containing strains and MRSA strains. The 50% effective doses (ED50s) for Streptococcus pneumoniae obtained ranged from 0.45 mg/kg to 3.39 mg/kg, the ED50s for Staphylococcus aureus obtained ranged from 0.30 mg/kg to 1.74 mg/kg, and the ED50 for Escherichia coli was 2.02 mg/kg. These results demonstrate potent in vivo efficacy including activity against strains containing common resistance determinants. Omadacycline demonstrated in vitro activity against a broad range of Gram-positive and select Gram-negative pathogens, including resistance determinant-containing strains, and this activity translated to potent efficacy in vivo.[2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.5320 mL | 7.6598 mL | 15.3196 mL | |
| 5 mM | 0.3064 mL | 1.5320 mL | 3.0639 mL | |
| 10 mM | 0.1532 mL | 0.7660 mL | 1.5320 mL |