Physicochemical Properties
| Molecular Formula | C26H32FN3O4 |
| Molecular Weight | 469.548390388489 |
| Exact Mass | 469.237 |
| CAS # | 312752-85-5 |
| PubChem CID | 9894205 |
| Appearance | White to off-white solid powder |
| Density | 1.2±0.1 g/cm3 |
| Boiling Point | 680.5±55.0 °C at 760 mmHg |
| Flash Point | 365.3±31.5 °C |
| Vapour Pressure | 0.0±2.1 mmHg at 25°C |
| Index of Refraction | 1.571 |
| LogP | 3.13 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 34 |
| Complexity | 687 |
| Defined Atom Stereocenter Count | 1 |
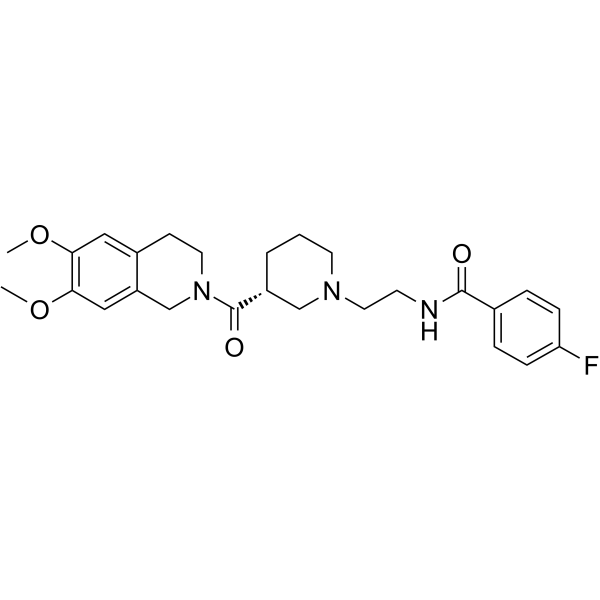
| SMILES | O=C([C@H]1CN(CCC1)CCNC(C2=CC=C(F)C=C2)=O)N3CC4=CC(OC)=C(OC)C=C4CC3 |
| InChi Key | MWLKUSHZNSYRKK-HXUWFJFHSA-N |
| InChi Code | InChI=1S/C26H32FN3O4/c1-33-23-14-19-9-12-30(17-21(19)15-24(23)34-2)26(32)20-4-3-11-29(16-20)13-10-28-25(31)18-5-7-22(27)8-6-18/h5-8,14-15,20H,3-4,9-13,16-17H2,1-2H3,(H,28,31)/t20-/m1/s1 |
| Chemical Name | N-[2-[(3R)-3-(6,7-dimethoxy-3,4-dihydro-1H-isoquinoline-2-carbonyl)piperidin-1-yl]ethyl]-4-fluorobenzamide |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
- "Funny" if current channel (pacemaker current channel) (IC50 = 0.12 μM for human HCN4 channel; IC50 = 0.09 μM for rat HCN4 channel)[2] |
| ln Vitro |
It was investigated how YM758 inhibited the rat/human organic cation transporter (hOCT1/rOct1)'s ability to absorb [3H]MPP. YM758 exhibits concentration-dependent inhibition of rOct1- and hOCT1-mediated [3H]MPP uptake, with IC50 values of 23.8 and 40.5 μM, respectively. Similar to the IC50 value for [3H]MPP uptake, the IC50 value for YM758 uptake of [14C]metformin via rOct1 can be estimated to be below 10 μM. Furthermore, YM758's inhibitory effect on [3H]E217βG uptake through OATP1B1 and OATP1B3 was investigated. With an IC50 value of 13.0 μM, YM758 inhibits [3H]E217βG uptake mediated by OATP1B1 in a concentration-dependent manner. The uptake of [3H]E217βG mediated by OATP1B3 is not inhibited by YM758 [1]. - Cardiac pacemaker cells (human/rat HCN4-transfected HEK293 cells, guinea pig sinoatrial node cells): YM758 selectively inhibits the "funny" if current in a concentration-dependent manner, reducing the amplitude of if current and slowing the spontaneous pacing rate of sinoatrial node cells[2] - Hepatocytes (rat and human): YM758 is actively taken up by hepatocytes via organic anion-transporting polypeptides (OATPs), with significant uptake observed in rat hepatocytes and moderate uptake in human hepatocytes[1] |
| ln Vivo |
YM758 plasma concentrations dropped quickly in beagles with tachycardia following a single intravenous dose of 0.03, 0.1, and 0.3 mg/kg; the corresponding t1/2 values were 1.62, 4.93, and 1.63 hours. The Vdss values were 3.19, 5.78, and 2.94 L/kg, while the CLtot values were 1.71, 1.69, and 1.48 L/h/kg at the corresponding doses. Greater t1/2 and Vdss values were obtained in comparison to other administration groups because the 0.1 mg/kg administration group only had blood drug concentration measured 24 hours after treatment. In dogs with tachycardia, the PK profile of YM758 seems to be linear between 0.03 and 0.3 mg/kg. On a blood basis (CLb, dog), the CLtot of YM758 is predicted to be 1.47 to 1.69 L/h/kg [2]. After administering 14C-YM758, extract radioactivity from rat eyeballs using a solution of 2mol/L hydrochloric acid and methanol (5:95, v/v); the radioactivity recovery rate was 67.1% after 24 hours and 97.1% after 4 hours. After 4 hours and 24 hours, respectively, the extracted samples' radioactive HPLC recoveries were 90.6% and 100.6%. Four hours after delivery, the primary molecule found in the eyes was YM758, the initial medication, accounting for 66.7% of the sample. Additionally, the metabolites YM-252124 (14.5%), YM-394111 (2.4%), and YM-234903 (1.8%) were noted [3]. - Tachycardia animal models (rats, dogs): Intravenous or oral administration of YM758 (0.3-3 mg/kg) produces a dose-dependent reduction in heart rate, with maximal heart rate reduction of 20-30% in tachycardic rats and dogs; the effect duration correlates with plasma drug concentration[2] - Rats: YM758 shows rapid hepatic uptake after intravenous administration, with high concentrations detected in liver tissue within 30 minutes; it is primarily excreted via bile, with minimal urinary excretion[1] - Humans: Single oral administration of YM758 (5-20 mg) results in dose-dependent plasma exposure, and heart rate reduction is positively correlated with plasma drug concentration (r = 0.86)[2] |
| Enzyme Assay |
- if current recording assay (patch-clamp technique): HCN4-transfected HEK293 cells or isolated guinea pig sinoatrial node cells are maintained in a recording chamber with physiological buffer. YM758 is added at gradient concentrations (0.01-1 μM) and incubated for 10 minutes. The if current is recorded using whole-cell patch-clamp configuration under voltage-clamp conditions (holding potential -40 mV, step depolarization to -120 mV). Current amplitude and decay kinetics are analyzed to calculate IC50 values[2] |
| Cell Assay |
- Hepatocellular uptake assay: Isolated rat or human hepatocytes are suspended in uptake buffer and incubated with YM758 (1-10 μM) at 37°C for 5-30 minutes. The reaction is terminated by adding ice-cold buffer, and cells are centrifuged to separate intracellular and extracellular drug. The concentration of YM758 in cell pellets is quantified by LC-MS/MS to evaluate uptake efficiency[1] - Pacing rate assay: Primary guinea pig sinoatrial node cells are cultured in serum-containing medium. YM758 (0.05-0.5 μM) is added, and spontaneous pacing rate is monitored for 60 minutes using a phase-contrast microscope coupled with a rate counter. The percentage change in pacing rate compared to baseline is calculated[2] |
| Animal Protocol |
- Rat pharmacokinetic (PK) and hepatic excretion study: Male rats are randomly divided into intravenous (i.v.) and oral (p.o.) administration groups. YM758 is dissolved in a mixture of ethanol and saline (1:9, v/v) and administered at a dose of 1 mg/kg (i.v.) or 5 mg/kg (p.o.). Blood samples are collected at 0.083, 0.25, 0.5, 1, 2, 4, 8, 24 hours post-administration. Bile and urine are collected for 24 hours via bile duct and bladder cannulation. Liver tissue is harvested at 2 hours post-i.v. administration. Drug concentrations in plasma, bile, urine, and liver are determined by LC-MS/MS[1] - Dog tachycardia model heart rate study: Beagle dogs with electrically induced tachycardia (heart rate > 180 bpm) are administered YM758 via intravenous infusion (0.1-1 mg/kg over 30 minutes). Heart rate is continuously monitored via ECG, and plasma drug concentrations are measured at 0, 30, 60, 120, 240 minutes post-infusion. The relationship between plasma concentration and heart rate reduction is analyzed[2] - Rat long-term retention study: Male rats are administered YM758 (1 mg/kg, i.v.) and sacrificed at 1, 3, 7, 14 days post-administration. Liver, kidney, and heart tissues are collected, and concentrations of unchanged YM758 and its metabolites are quantified by LC-MS/MS to evaluate tissue retention[3] |
| ADME/Pharmacokinetics |
- Absorption: Oral bioavailability in rats is approximately 23%[1] - Distribution: Rapidly distributed to liver (highest tissue concentration) in rats, with liver-to-plasma concentration ratio of 12.8 at 2 hours post-i.v. administration; moderate distribution to heart and kidney[1][3] - Metabolism: Undergoes minimal metabolism in rats and humans; major circulating form is unchanged YM758; two minor metabolites (hydroxylated derivatives) are detected in liver tissue, accounting for <10% of total drug-related material[3] - Excretion: Primarily excreted via bile in rats (78% of i.v. dose recovered in bile within 24 hours); urinary excretion is negligible (<5%)[1] - Pharmacokinetic parameters (rats, i.v. 1 mg/kg): Terminal half-life (t₁/₂) = 3.2 hours; volume of distribution (Vd) = 0.8 L/kg; total body clearance (CL) = 0.18 L/h/kg[1] - Pharmacokinetic parameters (humans, p.o. 10 mg): t₁/₂ = 4.5 hours; Cmax = 89 ng/mL; AUC₀-∞ = 420 ng·h/mL[1] |
| Toxicity/Toxicokinetics |
- Plasma protein binding: High plasma protein binding in rats (92-94%) and humans (95-97%)[1] - Hepatic safety: No obvious hepatotoxicity in rats after 14-day repeated administration (1 mg/kg/day, i.v.), as indicated by normal serum ALT, AST, and bilirubin levels[3] |
| References |
[1]. Umehara K, et al. Hepatic uptake and excretion of (-)-N-{2-[(R)-3-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-2-carbonyl)piperidino]ethyl}-4-fluorobenzamide (YM758), a novel if channel inhibitor, in rats and humans. Drug Metab Dispos. 2008 Ju [2]. Umehara K, et al. Relationship between exposure of (-)-N-{2-[(R)-3-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-2-carbonyl)piperidino]ethyl}-4-fluorobenzamide (YM758), a "funny" if current channel inhibitor, and heart rate reduction in tachyca [3]. Umehara K, et al. Investigation of long-term retention of unchanged (-)-N-{2-[(R)-3-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-2-carbonyl)piperidino]ethyl}-4-fluorobenzamide, a novel "funny" If current channel inhibitor, and its metabolites |
| Additional Infomation |
- YM758 is a novel, selective inhibitor of the "funny" if current (pacemaker current) expressed in cardiac sinoatrial node cells[2][3] - Its mechanism of action involves blocking the if current mediated by HCN4 channels, which reduces the spontaneous firing rate of sinoatrial node cells, thereby decreasing heart rate in tachycardic conditions[2] - The high liver uptake and biliary excretion contribute to its low systemic exposure and favorable safety profile[1] - It is being developed for the treatment of tachyarrhythmias due to its selective heart rate-lowering effect without affecting cardiac contractility[2] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~100 mg/mL (~212.97 mM) |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1297 mL | 10.6485 mL | 21.2970 mL | |
| 5 mM | 0.4259 mL | 2.1297 mL | 4.2594 mL | |
| 10 mM | 0.2130 mL | 1.0648 mL | 2.1297 mL |