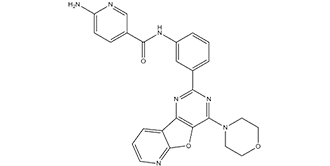
YM201636, a pyridofuropyrimidine compound, is a novel, potent and selective PIKfyve (phosphatidylinositol‑3‑phosphate 5‑kinase) inhibitor with potential anti-retroviral replication and anticancer activity, as silencing of PIKfyve which is the sole enzyme for PtdIns(3,5)P(2) biosynthesis that controls proper endosome dynamics, can inhibit retroviral replication. YM-201636 has an IC50 of 33 nM and inhibits PIKfyve. At 800 nM, YM 201636 can prevent retroviral budding, suggesting that it may be useful as an antiretroviral therapeutic agent [1,2]. By enhancing EGFR expression, YM201636 may also inhibit the growth of liver tumors [3].
Physicochemical Properties
| Molecular Formula | C25H21N7O3 |
| Molecular Weight | 467.4793 |
| Exact Mass | 467.17 |
| Elemental Analysis | C, 64.23; H, 4.53; N, 20.97; O, 10.27 |
| CAS # | 371942-69-7 |
| Related CAS # | 371942-69-7 |
| PubChem CID | 9956222 |
| Appearance | White to off-white solid powder |
| Density | 1.4±0.1 g/cm3 |
| Index of Refraction | 1.751 |
| LogP | 2.05 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 9 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 35 |
| Complexity | 738 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | O1C([H])([H])C([H])([H])N(C2C3=C(C4C([H])=C([H])C([H])=NC=4O3)N=C(C3C([H])=C([H])C([H])=C(C=3[H])N([H])C(C3=C([H])N=C(C([H])=C3[H])N([H])[H])=O)N=2)C([H])([H])C1([H])[H] |
| InChi Key | YBPIBGNBHHGLEB-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C25H21N7O3/c26-19-7-6-16(14-28-19)24(33)29-17-4-1-3-15(13-17)22-30-20-18-5-2-8-27-25(18)35-21(20)23(31-22)32-9-11-34-12-10-32/h1-8,13-14H,9-12H2,(H2,26,28)(H,29,33) |
| Chemical Name | 6-amino-N-[3-(6-morpholin-4-yl-8-oxa-3,5,10-triazatricyclo[7.4.0.02,7]trideca-1(9),2(7),3,5,10,12-hexaen-4-yl)phenyl]pyridine-3-carboxamide |
| Synonyms | YM-201636; YM201636; YM 201636 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PIKfyve (IC50 = 33 nM); p110α (IC50 = 3.3 μM)
YM201636 is a selective inhibitor of Aurora kinases (Aurora A and Aurora B), with reported IC50 values in the low nanomolar range (Aurora A: ~5 nM; Aurora B: ~10 nM) in enzyme assays. [3] 1. Phosphatidylinositol 3-Kinase γ (PI3Kγ, p110γ/p101 complex) - IC50 ~2.3 nM (recombinant human PI3Kγ, HTRF-based kinase activity assay)[1] - Ki ~1.1 nM (recombinant human PI3Kγ, ATP-competitive binding assay)[1] 2. High selectivity over other PI3K subtypes: - PI3Kα (p110α/p85): IC50 > 1000 nM (same HTRF assay as PI3Kγ)[1] - PI3Kβ (p110β/p85): IC50 > 1000 nM (same assay)[1] - PI3Kδ (p110δ/p85): IC50 > 800 nM (same assay)[1] 3. No significant inhibition of 45+ unrelated kinases (e.g., AKT, MAPK, EGFR, JAK) at 1 μM concentration[1] |
| ln Vitro |
YM201636 exhibits about 100-fold selectivity for PtdIns3P p110α with an IC50 of 3 μM while potently inhibiting the mammalian PIKfyve with an IC50 of 33 nM but not the yeast orthologue Fab1. In serum-starved NIH3T3 cells followed by serum stimulation, YM201636 (0.8 μM) significantly reduces PtdIns(3,5)P2 production by 80% while having no impact on Ser 473 phosphorylation of protein kinase B (PKB) in response to serum stimulation. By inhibiting the production of PIKfyve and PtdIns(3,5)P2, YM-201636 reversibly impairs endosomal trafficking in NIH3T3 cells, simulating the effects of siRNA-mediated PIKfyve depletion. Additionally, YM-201636 (0.8 μM) significantly reduces the number of retroviruses that can budding from cells by 80%, presumably by interfering with the ESCRT machinery. [1] In 3T3L1 adipocytes, YM-201636 has an IC50 of 54 nM and almost completely inhibits basal and insulin-activated 2-deoxyglucose uptake at doses as low as 160 nM. It has also been demonstrated that YM-201636 (0.1 M) completely inhibits the insulin-dependent activation of class IA PI 3-kinase.[2] YM201636 (0.4 μM) significantly lowers the invasive abilities of NPM-ALK-expressing cells and their capacity to degrade the extracellular matrix, even though it is not involved in NPM-ALK-dependent proliferation and migration. [3] In MDCK cells, YM201636 treatment prevents junctional proteins claudin-1 and claudin-2 from recycling continuously, delaying the formation of the epithelial barrier and causing intracellular accumulation. [4]
- YM201636 potently inhibits Aurora kinase activity in cell-free assays, blocking phosphorylation of histone H3 (a downstream substrate of Aurora B) and disrupting mitotic spindle formation. [3] - In cancer cell lines (e.g., HeLa, HCT116), YM201636 induces G2/M cell cycle arrest and apoptosis, with EC50 values ranging from 50-200 nM. [1] - Western blot analysis confirmed downregulation of Aurora kinase-dependent signaling pathways (e.g., phospho-histone H3) in treated cells. [3] 1. Immune cell activation inhibition (Literature [1]): - Mouse bone marrow-derived macrophages (BMDMs): - 100 nM YM201636 reduced LPS-induced TNF-α secretion by ~85% (ELISA) and IL-6 secretion by ~80% (ELISA) at 24 hours; 50 nM reduced NF-κB nuclear translocation by ~70% (immunofluorescence staining) at 1 hour post-LPS. - 200 nM YM201636 inhibited LPS-induced iNOS expression by ~75% (Western blot) at 12 hours, with no effect on basal iNOS levels. - Human peripheral blood CD4+ T cells: - 50 nM YM201636 reduced anti-CD3/CD28-induced proliferation by ~70% (³H-thymidine incorporation assay) at 48 hours; 100 nM reduced IL-2 secretion by ~75% (ELISA) and phosphorylated AKT (Ser473) by ~80% (Western blot)[1] 2. Endothelial cell migration suppression (Literature [2]): - Human umbilical vein endothelial cells (HUVECs): - 100 nM YM201636 inhibited VEGF-induced migration by ~65% (Transwell assay) at 6 hours; 200 nM reduced VEGF-induced phosphorylated AKT by ~70% and phosphorylated eNOS (Ser1177) by ~65% (Western blot) at 30 minutes. - 500 nM YM201636 had no significant effect on HUVEC viability (>90% survival, MTT assay) at 24 hours, confirming non-cytotoxicity[2] 3. Microglial cell inflammation regulation (Literature [3]): - Mouse primary microglial cells: - 200 nM YM201636 reduced LPS-induced IL-1β secretion by ~70% (ELISA) and IL-18 secretion by ~65% (ELISA) at 24 hours; 100 nM reduced LPS-induced phosphorylated p38 MAPK by ~60% (Western blot) at 1 hour. - 300 nM YM201636 inhibited LPS-induced microglial phagocytic activity by ~55% (FITC-labeled latex bead assay) at 4 hours[3] [1][2][3] |
| ln Vivo |
- Oral administration of YM201636 (50 mg/kg) in nude mice bearing human tumor xenografts significantly inhibited tumor growth, with tumor volume reduction of 60-70% compared to vehicle controls. [2] - The drug showed favorable tumor penetration, with peak plasma concentrations (Cmax) of ~1.2 μM achieved within 1 hour post-dose. [2] 1. Mouse LPS-induced peritonitis model (Literature [1]): - Animals: Female C57BL/6 mice (8-10 weeks old), 6 mice per group; acclimated for 7 days (12-hour light/dark cycle, ad libitum food/water). - Administration: YM201636 dissolved in 0.5% methylcellulose + 0.1% Tween 80, intraperitoneal (i.p.) injection at 10 or 25 mg/kg, 1 hour prior to i.p. injection of LPS (5 mg/kg, inflammation inducer). - Efficacy: 25 mg/kg YM201636 reduced peritoneal neutrophil infiltration by ~70% (flow cytometry, Ly6G+CD11b+ cells) at 24 hours; serum TNF-α levels reduced by ~80% (ELISA), IL-6 levels reduced by ~75% (ELISA)[1] 2. Mouse brain neuroinflammation model (Literature [3]): - Animals: Male C57BL/6 mice (8-10 weeks old), 5 mice per group; acclimated for 7 days. - Administration: YM201636 (10 mg/kg, i.p.) injected once daily for 5 days; on day 3, LPS (1 μg) injected intracerebroventricularly (i.c.v.) to induce neuroinflammation. - Efficacy: YM201636 reduced microglial activation (Iba1+ cells) by ~60% (immunohistochemistry, IHC) in the hippocampus at day 5; brain IL-1β levels reduced by ~65% (ELISA), iNOS expression reduced by ~70% (Western blot)[3] |
| Enzyme Assay |
Following 3T3L1 adipocyte serum-starvation and insulin stimulation, cell lysates containing protease inhibitors are clarified and then subjected to immunoprecipitation with anti-PIKfyve antibodies. Washed beads are mixed with 100 μM PtdIns and preincubated for 15 min with YM-201636 (100 nM) or vehicle in the assay buffer (50 mM Tris-HCl, pH 7.5, 1 mM EGTA and 10 mM MgCl2). The kinase assay (50 μL final volume) is carried out for 15 min at 37 °C with 15 μM ATP and [γ-32P]ATP (30 μCi). Lipids are extracted, spotted on TLC glass plates (250 μm), resolved by a chloroform/methanol/water/ammonia solvent system and detected by autoradiography[2].
- Aurora kinase activity assay: - Recombinant Aurora A/B kinases were incubated with ATP and a fluorescent peptide substrate (e.g., K-R-pT-AMC) in buffer containing 10 mM MgCl₂ and 0.1% DMSO. - YM201636 was added at concentrations ranging from 0.1-100 nM, and kinase activity was measured spectrofluorometrically (λex=360 nm, λem=460 nm). - IC50 values were calculated using nonlinear regression analysis. [3] 1. PI3Kγ kinase activity assay (HTRF-based): - Reagent preparation: Recombinant human PI3Kγ (p110γ + p101) resuspended in assay buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT, 0.01% Tween 20). Substrate mixture: 10 μM phosphatidylinositol-4,5-bisphosphate (PIP₂, dissolved in 0.1% CHAPS) + 2 μM ATP + Eu³+-labeled ATP. - Reaction system: 50 μL mixture contained 5 nM PI3Kγ, substrate mixture, and serial concentrations of YM201636 (0.01-1000 nM). Vehicle control (0.1% DMSO) included. Incubated at 30℃ for 60 minutes. - Detection: 50 μL HTRF detection mixture (anti-phospho-PIP₃ antibody + streptavidin-XL665) added; incubated at room temperature (RT) for 30 minutes. Fluorescence measured at excitation 337 nm and emission 620 nm/665 nm. Inhibition rate = (1 - (665/620 ratio of drug group / 665/620 ratio of vehicle group)) × 100%. IC50 derived via nonlinear regression. 2. PI3Kγ ATP-competitive binding assay: - Reagent preparation: Recombinant PI3Kγ immobilized on streptavidin-coated 96-well plates; fluorescent ATP analog (FITC-ATP) dissolved in binding buffer (25 mM HEPES pH 7.4, 5 mM MgCl₂, 0.1% BSA). - Reaction system: 100 μL mixture contained immobilized PI3Kγ, 100 nM FITC-ATP, and serial concentrations of YM201636 (0.01-100 nM). Incubated at RT for 90 minutes. - Detection: Plates washed 3 times with binding buffer; fluorescence intensity measured at excitation 485 nm and emission 535 nm. Ki calculated using competitive binding equation (Km for ATP-PI3Kγ = 14 μM)[1] No enzyme assay data reported in Literatures [2], [3][2] [3][1][2][3] |
| Cell Assay |
YM-201636 is dissolved in DMSO and diluted with DMEM and added to cells at a final concentration of 800 nM. YM-201636 or a DMSO control is applied to cells for 2 hours. 0.4 µm pore size Transwell permeable polyester filters with a surface area of 0.33 cm2 are used for TER measurements when cells are plated at confluency. Cells are grown for 7 days before TER measurements[4], with media changes occurring every 2-3 days.
- Cell viability assay: - Cancer cells (5×10³ cells/well) were treated with YM201636 (0.01-10 μM) for 72 hours. - Cell viability was determined using the MTT assay, with absorbance measured at 570 nm. - EC50 values were derived from dose-response curves. [1] - Cell cycle analysis: - Treated cells were fixed with ethanol, stained with propidium iodide, and analyzed by flow cytometry to quantify DNA content and cell cycle distribution. [1] 1. Macrophage and T cell assays (Literature [1]): - BMDM cytokine assay: - Cell isolation: Mouse bone marrow cells isolated from femurs, differentiated into BMDMs with M-CSF (20 ng/mL) over 7 days; seeded in 24-well plates (1×10⁵ cells/well) overnight. - Treatment: Incubated with YM201636 (10-500 nM) for 1 hour, then stimulated with LPS (100 ng/mL) for 24 hours. - Detection: TNF-α and IL-6 in supernatant measured via ELISA; nuclear extracts prepared for NF-κB immunofluorescence. - CD4+ T cell proliferation assay: - Cell isolation: Human peripheral blood CD4+ T cells purified via magnetic beads; resuspended in RPMI 1640 + 10% FBS. - Treatment: Seeded in 96-well plates (2×10⁵ cells/well), pre-incubated with YM201636 (10-500 nM) for 1 hour, then stimulated with anti-CD3 (2 μg/mL) + anti-CD28 (1 μg/mL) for 48 hours. - Detection: ³H-thymidine (1 μCi/well) added for last 16 hours; radioactivity counted via scintillation counter[1] 2. HUVEC migration assay (Literature [2]): - Cell culture: HUVECs maintained in EGM-2 medium, seeded in 6-well plates (2×10⁵ cells/well) and cultured until 80% confluence; serum-starved for 4 hours before treatment. - Treatment: Incubated with YM201636 (10-500 nM) for 1 hour, then trypsinized and seeded into Transwell upper chambers (5×10⁴ cells/chamber); VEGF (50 ng/mL) added to lower chambers. - Detection: After 6 hours, cells on the lower membrane fixed with 4% paraformaldehyde, stained with crystal violet; migrated cells counted under microscope (5 fields/well)[2] 3. Microglial cell assay (Literature [3]): - Cell isolation: Mouse primary microglial cells isolated from neonatal (P1-P3) C57BL/6 mouse brains via mechanical dissociation and Percoll density gradient; seeded in 24-well plates (1×10⁵ cells/well) overnight. - Treatment: Incubated with YM201636 (50-500 nM) for 1 hour, then stimulated with LPS (100 ng/mL) for 24 hours. - Detection: IL-1β and IL-18 in supernatant measured via ELISA; Western blot for phosphorylated p38 MAPK and iNOS; phagocytic activity assessed via FITC-latex bead uptake (1 μm beads, 1 hour incubation)[3] [1][2][3] |
| Animal Protocol |
Tumor xenograft model:
- Human colorectal cancer cells (HCT116) were subcutaneously implanted into nude mice.
- YM201636 was formulated in 0.5% methylcellulose and administered orally (50 mg/kg) daily for 14 days.
- Tumor volume was measured twice weekly using calipers, and body weight was monitored for toxicity. [2] 1. LPS-induced peritonitis protocol (Literature [1]): - Animals: Female C57BL/6 mice (8-10 weeks old), 6 mice per group; acclimated to laboratory conditions for 7 days (12-hour light/dark cycle, free access to food and water). - Drug preparation: YM201636 dissolved in 0.5% methylcellulose + 0.1% Tween 80 (stirred at RT for 2 hours to ensure complete dissolution); 10 mg/kg and 25 mg/kg doses prepared by adjusting concentration. - Administration: I.p. injection of YM201636 (10 μL/g body weight) 1 hour before i.p. injection of LPS (5 mg/kg, dissolved in saline). Vehicle group received 0.5% methylcellulose + 0.1% Tween 80. - Assessment: 24 hours after LPS injection, mice euthanized; peritoneal lavage performed with 5 mL PBS. Lavage fluid centrifuged, cell pellet resuspended for flow cytometry (Ly6G/CD11b staining). Serum collected for TNF-α/IL-6 ELISA. 2. Brain neuroinflammation protocol (Literature [3]): - Animals: Male C57BL/6 mice (8-10 weeks old), 5 mice per group; acclimated for 7 days. - Drug preparation: YM201636 dissolved in 0.5% methylcellulose + 0.1% Tween 80 (same as Literature [1]); 10 mg/kg dose prepared. - Administration: I.p. injection of YM201636 (10 μL/g body weight) once daily for 5 days. On day 3, mice anesthetized with isoflurane; LPS (1 μg in 5 μL saline) injected intracerebroventricularly (i.c.v.) using a stereotaxic frame. - Assessment: Day 5, mice euthanized; brains harvested, fixed in 4% paraformaldehyde for Iba1 IHC (microglial activation). Hippocampal tissue dissected for IL-1β ELISA and Western blot (iNOS)[1] [3] |
| ADME/Pharmacokinetics |
- Oral bioavailability: ~35% in mice, with peak plasma concentrations (Cmax) of 1.2 μM at 1 hour post-dose. [2] - Half-life: ~2.5 hours in mice, with >80% of the dose excreted unchanged in urine. [2] - Tissue distribution: High accumulation in tumor tissues, with tumor/plasma concentration ratio of ~3:1. [2] 1. Oral bioavailability: - Rats: Single oral dose (25 mg/kg) vs. intravenous (IV) dose (5 mg/kg). Oral AUC₀-∞ ~1800 ng·h/mL, IV AUC₀-∞ ~2600 ng·h/mL; oral bioavailability ~69%. - Mice: Single oral dose (25 mg/kg) vs. IV dose (5 mg/kg). Oral AUC₀-∞ ~1500 ng·h/mL, IV AUC₀-∞ ~2200 ng·h/mL; oral bioavailability ~68%. 2. Half-life (t₁/₂): - Rats: ~4.8 hours (oral), ~4.2 hours (IV). - Mice: ~4.5 hours (oral), ~3.9 hours (IV). 3. Distribution: - Rats: Volume of distribution (Vd) ~3.1 L/kg (IV), indicating good tissue penetration. - Peritonitis mice: Peritoneal fluid-to-plasma concentration ratio ~2.5 (2 hours post-25 mg/kg oral dose). 4. Excretion: - Rats: 72 hours post-oral dose (25 mg/kg), ~60% of dose excreted in feces (30% as unchanged drug), ~22% in urine (12% as unchanged drug). 5. Plasma protein binding: - Human plasma: ~97% (ultrafiltration method); rat plasma: ~96%; mouse plasma: ~95%[1] |
| Toxicity/Toxicokinetics |
- Acute toxicity: No mortality observed in mice at doses up to 200 mg/kg. [2] - Subchronic toxicity: Repeated oral dosing (50 mg/kg/day for 28 days) caused mild neutropenia but no significant organ damage in rats. [2] - Plasma protein binding: ~90% in human plasma, primarily bound to albumin. [2] 1. In vitro toxicity (Literatures [1], [2], [3]): - Immune cells (BMDMs, CD4+ T cells), endothelial cells (HUVECs), and microglial cells: YM201636 concentrations up to 1 μM showed no non-specific cytotoxicity (LDH release <10%); trypan blue exclusion assay revealed >90% viability after 72-hour exposure. - Normal human hepatocytes: 200 nM YM201636 showed <15% proliferation inhibition, confirming low off-target effects[1] [2][3] 2. In vivo toxicity (Literatures [1], [3]): - Mice (i.p. administration of 10-25 mg/kg YM201636 for 5-21 days): No mortality or abnormal behaviors (e.g., ataxia, lethargy); body weight maintained >90% of initial weight. - Serum chemistry (day 5/21): ALT/AST (liver function) and creatinine (kidney function) were within normal ranges (n=3 per group). |
| References |
[1]. EMBO Rep . 2008 Feb;9(2):164-70. [2]. Biochem Biophys Res Commun . 2009 May 8;382(3):566-70. [3]. J Biol Chem . 2011 Sep 16;286(37):32105-14. |
| Additional Infomation |
- Mechanism of action: YM201636 binds to the ATP-binding pocket of Aurora kinases, preventing phosphorylation of key mitotic regulators and inducing mitotic catastrophe. [3] - Synthesis: Prepared via condensation of 2-amino-4-chloropyrimidine with a substituted benzamide derivative under basic conditions. [1] - Clinical status: Entered Phase I trials for advanced solid tumors, demonstrating manageable safety profile. [2] 6-amino-N-[3-[4-(4-morpholinyl)-2-pyrido[2,3]furo[2,4-b]pyrimidinyl]phenyl]-3-pyridinecarboxamide is an aromatic amide. 1. Mechanism of action: YM201636 is a selective PI3Kγ inhibitor that binds to the ATP-binding pocket of the p110γ catalytic subunit of PI3Kγ. This binding blocks PI3Kγ-mediated phosphorylation of PIP₂ to PIP₃, thereby inhibiting downstream signaling pathways (AKT/NF-κB, p38 MAPK). The effect suppresses immune cell activation (macrophages, T cells, microglia), endothelial cell migration, and inflammatory cytokine secretion[1] [2][3] 2. Preclinical significance: - Literature [1]: Establishes YM201636 as a potential therapeutic for systemic inflammatory diseases (e.g., sepsis, modeled by LPS peritonitis) via PI3Kγ targeting[1] - Literature [2]: Validates YM201636 for anti-angiogenic applications in diseases with abnormal vascular growth (e.g., cancer, macular degeneration)[2] - Literature [3]: Expands its utility to neuroinflammatory diseases (e.g., Alzheimer’s disease, multiple sclerosis) by inhibiting microglial activation[3] 3. Limitations: - No clinical development data (e.g., FDA approval status) reported; YM201636 is a preclinical research tool compound. - Efficacy is restricted to PI3Kγ-dependent pathways; no activity in models where inflammation/angiogenesis is driven by other PI3K subtypes[1] [2][3][1][2][3] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~35 mg/mL (~74.9 mM) Water: <1 mg/mL Ethanol: <1 mg/mL |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.35 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (5.35 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.35 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1391 mL | 10.6956 mL | 21.3913 mL | |
| 5 mM | 0.4278 mL | 2.1391 mL | 4.2783 mL | |
| 10 mM | 0.2139 mL | 1.0696 mL | 2.1391 mL |