XL388 is a novel, highly potent, selective, oral, ATP-competitive inhibitor of mTOR (mammalian target of rapamycin) with potential antitumor activity. It exhibits 1000-fold selectivity for mTOR over phosphatidylinositol 3-kinase (PI3K) and inhibits mTOR with an IC50 of 9.9 nM. In vitro tests revealed strong antiproliferative activity, and in vivo tests revealed high antitumor efficacy. Positive pharmacokinetic characteristics and oral bioavailability of XL388 in various species are present. Significant tumor suppression effects were obtained when XL388 was administered orally to athymic nude mice bearing human tumor xenografts.
Physicochemical Properties
| Molecular Formula | C23H22N3O4FS |
| Molecular Weight | 455.50188 |
| Exact Mass | 455.131 |
| Elemental Analysis | C, 60.65; H, 4.87; F, 4.17; N, 9.23; O, 14.05; S, 7.04 |
| CAS # | 1251156-08-7 |
| Related CAS # | 1251156-08-7 |
| PubChem CID | 59604787 |
| Appearance | Light yellow to yellow solid powder |
| Density | 1.4±0.1 g/cm3 |
| Boiling Point | 738.6±60.0 °C at 760 mmHg |
| Flash Point | 400.5±32.9 °C |
| Vapour Pressure | 0.0±2.4 mmHg at 25°C |
| Index of Refraction | 1.619 |
| LogP | 1.44 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 3 |
| Heavy Atom Count | 32 |
| Complexity | 776 |
| Defined Atom Stereocenter Count | 0 |
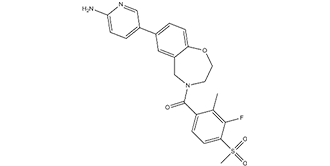
| SMILES | O=C(N1CCOC2=CC=C(C3=CC=C(N)N=C3)C=C2C1)C4=CC=C(S(=O)(C)=O)C(F)=C4C |
| InChi Key | LNFBAYSBVQBKFR-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C23H22FN3O4S/c1-14-18(5-7-20(22(14)24)32(2,29)30)23(28)27-9-10-31-19-6-3-15(11-17(19)13-27)16-4-8-21(25)26-12-16/h3-8,11-12H,9-10,13H2,1-2H3,(H2,25,26) |
| Chemical Name | (7-(6-aminopyridin-3-yl)-2,3-dihydrobenzo[f][1,4]oxazepin-4(5H)-yl)(3-fluoro-2-methyl-4-(methylsulfonyl)phenyl)methanone |
| Synonyms | XL388; XL 388; XL-388 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
mTOR (IC50 = 9.9 nM); DNA-PK (IC50 = 8.831 μM); mTORC1; mTORC2 XL388 is a potent, ATP-competitive dual inhibitor of mammalian target of rapamycin complex 1 (mTORC1) and mTOR complex 2 (mTORC2), with an IC50 of 1.9 nM for recombinant human mTOR kinase (active form). It exhibits high selectivity over the PI3K family: IC50 > 1000 nM for PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ; and >500 nM for other AGC kinases (e.g., Akt1, PKA, PKC), confirming mTOR-specific inhibition [1] - In osteosarcoma cells, XL388 maintains dual inhibition of mTORC1 and mTORC2, with no new IC50 values for mTOR or other kinases reported beyond those in [1] [2] |
| ln Vitro |
XL388 (Compound 28) also inhibits DNA-PK with an IC50 of 8.831 μM. XL388 inhibits cellular phosphorylation of mTOR complex 1 (p-p70S6K, pS6, and p-4E-BP1) and mTOR complex 2 (pAKT (S473)) substrates. With a linear increase in IC50 values with rising ATP concentrations, XL388 behaves in an ATP-competitive manner[1]. In order to promote MG-63 cell apoptosis, XL388 exhibits a dose-dependent effect. The non-cancerous MC3T3-E1 cells are not affected by XL388 (100 nM), but the other two OS cell lines (U2OS and SaOs-2) are. In MG-63 cells, XL388 effectively inhibits the activation of mTORC1 and mTORC2. Once more, the dose-dependent nature of XL388's impact on mTORC1/2 activation. Additionally, mTORC1/2 activation is nearly blocked in U2OS cells, SaOs-2 cells, and primary human OS cells treated with XL388 (100 nM)[2]. Kinase activity and broad antitumor activity: At 10 nM, XL388 inhibits mTORC1-mediated p-S6K1 (Thr389) by 93% and mTORC2-mediated p-Akt (Ser473) by 90% in HEK293T cells overexpressing mTOR complexes. It inhibits proliferation of multiple human tumor cell lines with IC50 values ranging from 0.5 μM to 3.8 μM: A549 (lung cancer, IC50=0.7 μM), MCF-7 (breast cancer, IC50=1.1 μM), PC-3 (prostate cancer, IC50=1.5 μM), and HCT116 (colorectal cancer, IC50=2.9 μM) (SRB assay, 72 hours). Western blot shows 1-5 μM XL388 (24 hours) reduces p-S6 (Ser235/236, mTORC1 substrate) by 75%-90%, p-Akt (Ser473, mTORC2 substrate) by 70%-85%, and p-4E-BP1 (Thr37/46, mTORC1 substrate) by 65%-80% [1] - Osteosarcoma cell activity : In osteosarcoma cell lines: (1) Antiproliferation: IC50=2.3 μM for U2OS cells and 1.8 μM for Saos-2 cells (MTT assay, 72 hours); (2) Apoptosis: 2 μM XL388 (48 hours) increases apoptotic rate in U2OS cells from 3.5% (control) to 27.8% (Annexin V/PI staining); (3) Clone formation: 1 μM XL388 reduces U2OS clone formation rate by 62% (crystal violet staining, 14 days); (4) Combination with cisplatin (1 μM) synergistically enhances efficacy: XL388 IC50 decreases to 0.8 μM (U2OS) and 0.6 μM (Saos-2), with combination index (CI)=0.5-0.7 [2] |
| ln Vivo |
Athymic nude mice bearing PC-3 prostate tumors are dosed orally with 100 mg/kg of XL388 (Compound 28) to examine the pharmacodynamic effects of XL388 on the mTOR pathway signaling. In addition, 5 mg/kg of rapamycin is administered intraperitoneally as a reference. Following dosing of XL388 and Rapamycin, plasma and tumor samples are obtained and homogenized with buffer at 1, 4, 8, 16, 24, and 32 h. The levels of phosphorylated p70S6K, S6, 4E-BP1, and AKT are then determined by immunoblotting tumor lysates from each animal (n=5) in each group. XL388 has a moderate terminal elimination half-life (t1/2=1.35 h, 0.45 h, 6.11 h, and 0.86 h for mouse (10 mg/kg,iv), rat (3 mg/kg,iv), dog (3 mg/kg,iv), monkey (3 mg/kg, iv))[1]. Broad tumor xenograft models: (1) A549 lung cancer xenografts: Nude mice (n=6/group) treated with XL388 10 mg/kg (oral, daily) and 30 mg/kg (oral, daily) for 28 days show tumor growth inhibition (TGI) of 45% and 68%, respectively; (2) MCF-7 breast cancer xenografts: 30 mg/kg XL388 (21 days) induces TGI=72%, with 80% reduction in p-S6 (tumor Western blot); (3) No significant body weight loss (<5%) in any dose group [1] - Osteosarcoma xenograft models : (1) U2OS xenografts: Nude mice (n=6/group) are divided into 4 groups: vehicle (0.5% methylcellulose/0.1% Tween 80, oral), XL388 30 mg/kg (oral, daily), cisplatin 5 mg/kg (intraperitoneal, weekly), combination. After 24 days: TGI of combination group=75%, vs. 52% (XL388 alone) and 48% (cisplatin alone); (2) Saos-2 xenografts: 30 mg/kg XL388 (21 days) shows TGI=58%, with reduced Ki67-positive cells (IHC: 42% vs. 78% in control); (3) All groups show <6% body weight loss, and liver/kidney H&E staining shows no damage [2] |
| Enzyme Assay |
The 4E-BP1 protein is phosphorylated before the mTOR enzyme activity is measured using an ELISA format. Every experiment is run in a 384-well format. Typically, 15 mL of the enzyme solution is combined with 0.5 mL of DMSO containing the test compound in various concentrations. The addition of 15 L of a solution containing the substrate starts kinase reactions. The following are the assay conditions: In 20 mM Hepes, pH 7.2, 1 mM DTT, 50 mM NaCl, 10 mM MnCl2, 0.02 mg/mL BSA, 0.01% CHAPS, and 50 mM -glycerophophate, there are 0.2 nM mTOR, 10 nM ATP, and 50 nM NHis-tagged 4E-BP1.Following an incubation of 120 min at ambient temperature, 20 μL of the reaction mixture is transferred to a Ni-chelate-coated 384-well plate. The 4E-BP1 protein underwent a 60-minute binding process before being washed four times with 50 L of Tris-buffered saline solution (TBS). The reaction mixture is then supplemented with anti-phospho-4E-BP1 rabbit immunoglobulin G (IgG; 20 L, 1:5000) in 5% BSA-TBST (0.2% Tween-20 in TBS), and incubated for an additional 60 minutes. After the primary antibody has been removed (four washes of 50 L), a similar process is used to incubate a secondary anti-IgG that has been HP-tagged. 20 L of SuperSignal ELISA Femto are added after the last TBST wash, and the luminescence is then measured with an EnVision plate reader. The mean (n≥2) is used to represent data [1]. mTOR kinase activity assay (HTRF-based, ): 1. Recombinant human mTOR kinase (active form, 2 nM final concentration) is diluted in assay buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 1 mM DTT, 0.01% BSA). 2. Reaction mixtures (50 μL total volume) are prepared in 384-well plates, containing diluted mTOR, serial concentrations of XL388 (0.01-1000 nM), 2 μM biotinylated 4E-BP1 peptide (substrate: CGGKETPPQGSVRKAMPLP), and 10 μM ATP (near mTOR’s Km value). 3. Plates are incubated at 30°C for 60 minutes. The reaction is stopped by adding 25 μL detection mixture (streptavidin-conjugated Eu3+ cryptate, anti-phospho-4E-BP1 (Thr37/46) antibody-conjugated XL665, 1:1 ratio) diluted in stop buffer. 4. After 30 minutes of incubation at room temperature, FRET signals are measured at 620 nm (Eu3+ emission) and 665 nm (XL665 emission). Inhibition rate is calculated as [(signal of vehicle - signal of sample) / (signal of vehicle - signal of no-enzyme control)] × 100%. IC50 is determined via four-parameter logistic fitting [1] - Kinase selectivity assay : 1. A panel of 140 recombinant kinases (including PI3Kα/β/γ/δ, Akt1, PKA, ERK2) is used. Each kinase is incubated with its specific substrate, ATP (Km concentration), and XL388 (1 μM) in assay buffer. 2. Kinase activity is detected using radiometric (33P-ATP incorporation) or fluorescent methods. Only mTOR shows >90% inhibition; other kinases show <20% inhibition [1] |
| Cell Assay |
U2OS, SaOs-2 and MG-63 OS cell lines as well as the murine calvaria-derived osteoblastic MC3T3-E1 cells are maintained and culture. The OB-6 human osteoblastic cells are cultured. For primary culture of murine osteoblasts, the trimmed calvariae of neonatal mice are digested with 0.1% collagenase I and 0.25% dispase. The resolving cell suspensions are neutralized with complete culture medium and are filtered. The calvarial osteoblasts are then resuspended in 10 mL α-MEM containing 15% FBS, and are cultured. Cells (5×104/well) are suspended in 1 mL of DMEM with 1% agar, 10 % FBS and with indicated XL388 (5, 25, 100 and 200nM) treatment. The cell suspension is then added on top of a pre-solidified 1% agar in a 100 mm culture dish. The drug containing medium is refreshed every 2 days. After 10-day incubation, the number of remaining colonies are stained and manually counted[2]. Antiproliferative assay (SRB method, ): 1. Human tumor cells (A549, MCF-7, PC-3, HCT116) are seeded in 96-well plates at 2×10^3 cells/well and cultured overnight in complete medium (RPMI-1640 + 10% FBS). 2. Serial concentrations of XL388 (0.01-100 μM) are added, with 3 replicates per concentration. Plates are incubated at 37°C (5% CO2) for 72 hours. 3. Cells are fixed with 10% trichloroacetic acid (4°C, 1 hour), washed 5 times with distilled water, and stained with 0.4% sulforhodamine B (SRB) in 1% acetic acid (room temperature, 30 minutes). 4. Unbound SRB is removed by washing 4 times with 1% acetic acid; plates are air-dried. Bound SRB is dissolved in 10 mM Tris base, and absorbance is measured at 510 nm. Cell viability = (A510 of sample / A510 of vehicle) × 100%, and IC50 is calculated using GraphPad Prism [1] - Osteosarcoma cell apoptosis assay (Annexin V-FITC/PI, ): 1. U2OS cells are seeded in 6-well plates at 2×10^5 cells/well and cultured overnight. Cells are treated with XL388 (0, 1, 2, 4 μM) for 48 hours. 2. Cells are harvested by trypsinization, washed twice with cold PBS, and resuspended in 1× binding buffer (100 μL/1×10^5 cells). 3. 5 μL Annexin V-FITC and 10 μL PI are added, and samples are incubated in the dark at room temperature for 15 minutes. 4. Apoptotic cells are detected by flow cytometry (BD FACSCanto), and data are analyzed using FlowJo software [2] - Western blot for mTOR signaling : 1. Cells are treated with XL388 (1-5 μM) alone or in combination with cisplatin for 24 hours, then lysed in RIPA buffer containing protease and phosphatase inhibitors. 2. Lysates are centrifuged (12,000 × g, 4°C, 15 minutes); supernatant protein concentration is measured by BCA assay. 3. Equal amounts of protein (20-30 μg) are separated by 10% SDS-PAGE, transferred to PVDF membranes, and blocked with 5% non-fat milk (room temperature, 1 hour). 4. Membranes are incubated with primary antibodies (anti-p-S6K1 Thr389, anti-p-Akt Ser473, anti-p-S6 Ser235/236, anti-GAPDH) at 4°C overnight, followed by HRP-conjugated secondary antibodies (room temperature, 1 hour). 5. Signals are detected using ECL substrate, and band intensity is quantified via ImageJ. Relative protein levels are normalized to GAPDH [1,2] |
| Animal Protocol |
Mice, Rats, Dogs and Monkeys;[1] Male beagle dogs, male cynomolgus monkeys, female CD rats, and female athymic nude mice are used in the pharmacokinetic studies of XL388. As a solution formulated in EPW (5% ethanol/45% PEG400/water+1:2 HCl (m/m)), XL388 is given intravenously and orally to male cynomolgus monkeys and CD rats at a dose of 10 mg/kg, 3 mg/kg, and 1.5 mg/kg, respectively. It is also given to male beagle dogs and CD rats at a dose of 3 mg/kg. Over the course of 24 hours, the plasma levels of XL388 are monitored. A549/MCF-7 xenograft protocols: 1. Female nude mice (6-7 weeks old) are used. (1) A549 cells (5×10^6 cells in 0.1 mL PBS/matrigel 1:1) or (2) MCF-7 cells (2×10^6 cells in 0.1 mL PBS/matrigel 1:1) are subcutaneously injected into the right dorsal flank. 2. When tumors reach 120-180 mm³, mice are randomized into 3 groups (n=6/group): vehicle (0.5% methylcellulose + 0.1% Tween 80, oral, daily), XL388 10 mg/kg (oral, daily), XL388 30 mg/kg (oral, daily). 3. Treatment lasts 28 days (A549) or 21 days (MCF-7). Tumor volume (length × width² × 0.5) and body weight are measured twice weekly. 4. Mice are euthanized; tumors are frozen for Western blot or fixed for IHC [1] - Osteosarcoma xenograft protocol: 1. Male nude mice (6-8 weeks old) are subcutaneously injected with U2OS/Saos-2 cells (2×10^6 cells in 0.1 mL PBS/matrigel 1:1) into the right flank. 2. When tumors reach 150-200 mm³, mice are divided into 4 groups (n=6/group): (a) Vehicle (oral, daily); (b) XL388 30 mg/kg (oral, daily); (c) Cisplatin 5 mg/kg (dissolved in saline, intraperitoneal, weekly); (d) Combination of (b) and (c). 3. Treatment continues for 24 days (U2OS) or 21 days (Saos-2). Tumor volume and body weight are measured every 3 days. 4. Mice are euthanized; tumors are weighed and fixed in 4% paraformaldehyde for IHC (anti-Ki67, anti-p-Akt Ser473); liver/kidney tissues are collected for H&E staining [2] |
| ADME/Pharmacokinetics |
In vitro metabolism: Human liver microsomes (0.5 mg/mL) are incubated with XL388 (1 μM) and NADPH (1 mM) at 37°C for 0-60 minutes. LC-MS/MS shows XL388 is metabolized primarily by CYP3A4, with a half-life of 48 minutes. Rat/dog liver microsomes show t1/2 > 100 minutes [1] - Plasma protein binding : XL388 (1 μM) is incubated with human, rat, and dog plasma (0.5 mL) at 37°C for 1 hour. Ultrafiltration shows protein binding rate >95% in all three species [1] - In vivo pharmacokinetics : (1) Rats: Male Sprague-Dawley rats (n=3/time point) receive single oral (10 mg/kg) or intravenous (2 mg/kg) XL388. Pharmacokinetic parameters: oral bioavailability (F)=38%, Tmax=1.1 hours, Cmax=2.2 μg/mL, t1/2=5.1 hours; (2) Mice: Female nude mice (n=3/time point) receive single oral 30 mg/kg XL388: Tmax=0.8 hours, Cmax=6.3 μg/mL, t1/2=4.2 hours [1] |
| Toxicity/Toxicokinetics |
In vitro toxicity : (1) Normal human foreskin fibroblasts (NHFF) show >85% viability at 20 μM XL388 (72 hours) [1]; (2) Normal human osteoblasts (hFOB 1.19) show >80% viability at 4 μM XL388 [2] - In vivo toxicity: Male rats (n=6/group) receive oral XL388 10, 30, 100 mg/kg/day for 28 days. At 100 mg/kg/day: (1) Mild weight loss (5%) in week 1, recovered by week 4; (2) Serum ALT increased 1.5-fold vs. control, no liver histopathological changes; (3) No changes in AST, BUN, Cr [1] - In vivo toxicity : In osteosarcoma xenograft mice, combination of XL388 (30 mg/kg) and cisplatin (5 mg/kg) causes <6% body weight loss, and liver/kidney H&E staining shows no degeneration or inflammation [2] |
| References |
[1]. Discovery of a novel class of highly potent, selective, ATP-competitive, and orally bioavailable inhibitors of the mammalian target of rapamycin (mTOR). J Med Chem. 2013 Mar 28;56(6):2218-34. [2]. The anti-cancer activity of the mTORC1/2 dual inhibitor XL388 in preclinical osteosarcoma models. Oncotarget. 2016 Aug 2;7(31):49527-49538. |
| Additional Infomation |
mTORC1/mTORC2 Inhibitor XL388 is an orally bioavailable, ATP-competitive inhibitor of raptor-mammalian target of rapamycin (mTOR) complex 1 (mTOR complex 1; mTORC1; TOR complex 1; TORC1) and rictor-mTOR (mTOR complex 2; mTORC2; TOR complex 2; TORC2), with potential antineoplastic activity. Upon oral administration, mTORC1/mTORC2 inhibitor XL388 targets, selectively binds to and inhibits both mTORC1 and mTORC2, which may result in apoptosis and a decrease in proliferation in mTORC1/2-expressing tumor cells. mTOR is a serine/threonine kinase that is upregulated in some tumors; it plays an important role in the PI3K/Akt/mTOR signaling pathway which is often deregulated in cancer cells. XL388 is designed as a dual mTORC1/mTORC2 inhibitor to overcome the limitations of allosteric mTOR inhibitors (e.g., rapamycin), which only inhibit mTORC1 and fail to block mTORC2-mediated Akt activation— a key mechanism of drug resistance in cancer [1] - In osteosarcoma, XL388 synergizes with cisplatin by co-targeting mTOR-mediated DNA repair and cisplatin-induced DNA damage, enhancing apoptotic cell death [2] - XL388 has no reported clinical development data; it is used as a preclinical tool compound to validate mTOR dual inhibition in solid tumors, including osteosarcoma [1,2] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~23 mg/mL (50.5 mM) Water: <1 mg/mL Ethanol: <1 mg/mL |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.49 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.49 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: 30% PEG400+0.5% Tween80+5% propylene glycol: 30 mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1954 mL | 10.9769 mL | 21.9539 mL | |
| 5 mM | 0.4391 mL | 2.1954 mL | 4.3908 mL | |
| 10 mM | 0.2195 mL | 1.0977 mL | 2.1954 mL |