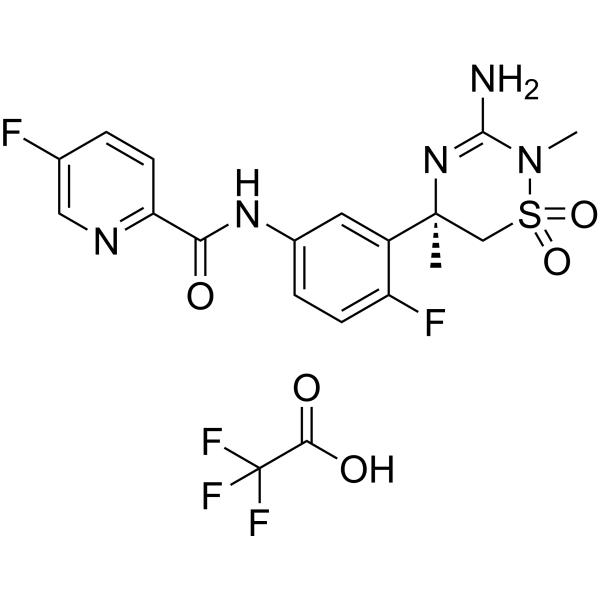
Verubecestat TFA (also known as MK-8931; SCH-900931), the trifluoroacetic acid salt form of MK-8931, is a beta-secretase 1 and BACE1 inhibitor with anti-AD (Alzheimer's disease) activity. It is in Phase 3 clinical trial for treatment of Alzheimer's disease. Amyloidogenic pathway in Alzheimer's disease (AD) involves breakdown of APP by β-secretase followed by γ-secretase and results in formation of amyloid beta plaque. β-secretase has been a promising target for developing novel anti-Alzheimer drugs.MK-8931 binds significantly to β-secretase. target: BACE1. In vitro:MK-8931 can effectively reduce Aβ40 in cells with a Ki of 7.8 nM and an IC50 of 13 nM. Docking revealed that, with respect to their free binding energy, acylguanidine 7a has the lowest binding energy followed by MK-8931 and pioglitazone and binds significantly to β-secretase.
Physicochemical Properties
| Molecular Formula | C19H18F5N5O5 | |
| Molecular Weight | 523.43 | |
| Exact Mass | 523.094 | |
| Elemental Analysis | C, 43.60; H, 3.47; F, 18.15; N, 13.38; O, 15.28; S, 6.12 | |
| CAS # | 2095432-65-6 | |
| Related CAS # | Verubecestat;1286770-55-5 | |
| PubChem CID | 129896720 | |
| Appearance | White to off-white solid powder | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 12 | |
| Rotatable Bond Count | 3 | |
| Heavy Atom Count | 35 | |
| Complexity | 827 | |
| Defined Atom Stereocenter Count | 1 | |
| SMILES | S1(C[C@@](C)(C2C(=CC=C(C=2)NC(C2C=CC(=CN=2)F)=O)F)N=C(N)N1C)(=O)=O.FC(C(=O)O)(F)F |
|
| InChi Key | MNYVOIVLGITLBF-LMOVPXPDSA-N | |
| InChi Code | InChI=1S/C17H17F2N5O3S.C2HF3O2/c1-17(9-28(26,27)24(2)16(20)23-17)12-7-11(4-5-13(12)19)22-15(25)14-6-3-10(18)8-21-14;3-2(4,5)1(6)7/h3-8H,9H2,1-2H3,(H2,20,23)(H,22,25);(H,6,7)/t17-;/m0./s1 | |
| Chemical Name | N-[3-[(5R)-3-amino-2,5-dimethyl-1,1-dioxo-6H-1,2,4-thiadiazin-5-yl]-4-fluorophenyl]-5-fluoropyridine-2-carboxamide;2,2,2-trifluoroacetic acid | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Verubecestat TFA (corresponding free base: Verubecestat, MK-8931) targets β-site amyloid precursor protein cleaving enzyme 1 (BACE1) with a Ki value of 0.4 nM (recombinant human BACE1) and an IC50 of 0.3 nM (enzymatic assay) [2] Verubecestat TFA (corresponding free base: Verubecestat, MK-8931) shows weak affinity for BACE2 with an IC50 of 450 nM, demonstrating >1500-fold selectivity for BACE1 over BACE2 [2] |
| ln Vitro |
An inhibitor of beta-site amyloid precursor protein cleaving enzyme 1/2 (BACE1/2) is called verubecestat TFA (MK-8931). Verubecestat TFA is unlikely to be an inhibitor of CYP-mediated drug-drug interactions because it does not significantly inhibit human CYP isoforms 1A2, 2C9, 2C19, 2D6, and 3A4 (IC50 >40 μM) [1]. Verubecestat TFA's IC50 values in HEK293 APPSwe/Lon cells for Aβ1-40, Aβ1-42, and sAPPβ are 2.1 nM, 0.7 nM, and 4.4 nM, respectively[1]. In recombinant human BACE1 enzymatic assay, the free base of Verubecestat TFA (Verubecestat) inhibited BACE1 activity in a dose-dependent manner with an IC50 of 0.3 nM; it did not significantly inhibit other aspartyl proteases (e.g., cathepsin D, renin) at concentrations up to 10 μM [2] - In human neuroblastoma SH-SY5Y cells stably expressing human APP695, the free base of Verubecestat TFA (Verubecestat) reduced Aβ40 and Aβ42 secretion with IC50 values of 1.2 nM and 1.5 nM, respectively [2] - In primary rat cortical neurons, the free base of Verubecestat TFA (Verubecestat) (1-100 nM) dose-dependently decreased Aβ40 levels in the culture medium, achieving 85% inhibition at 10 nM [2] - The free base of Verubecestat TFA (Verubecestat) showed no cytotoxicity in SH-SY5Y cells or primary cortical neurons at concentrations up to 10 μM after 72-hour incubation [2] |
| ln Vivo |
In Sprague-Dawley (SD) rats, verubecestat TFA (MK-8931; 3 mg/kg; IV or PO) has a T1/2 of 1.9 hours, a CL of 46 mL/min/kg, and a Vss of 5.4 L. AUC is 1.1 μM·h, C max is 0.27 μM, and /kg[1]. In cynomolgus monkeys, verubecestat TFA (1 mg/kg; IV) has a T1/2 of 4.9 hours, a CL of 21 mL/min/kg, and a Vss of 7.5 L/kg [1]. In beagle dogs, the T1/2 for verubecestat TFA (1 mg/kg; IV) is 9.7 hours, the CL is 4.3 mL/min/kg, and the Vss is 2.7 L/kg [1]. Rats treated with verubecestat TFA (30 mg/kg; PO; BID, 5 days) exhibit a moderate (1.4-fold) increase in CYP 3A1 activity, but CYP 1A1, 1A2, 2B, 3A2, and 4A expression remain largely unchanged[1]. Verubecestat TFA decreases CSF and cortical Aβ40 in a dose-dependent manner; its ED50 values are 5 and 8 mg/kg, respectively, and its EC50 values are 48 and 81 nM, respectively, for unbound plasma CSF and cortical Aβ40 [1]. The oral doses of verubecestat TFA (3 and 10 mg/kg) resulted in a significant decrease in CSF Aβ40 levels. The effect of the drug peaked 12 hours after dosing, with 72% and 81% reduction in CSF Aβ, respectively. %)[1]. In APP/PS1 transgenic mice (6 months old), oral administration of the free base of Verubecestat TFA (Verubecestat) (1, 3, or 10 mg/kg once daily for 28 days) dose-dependently reduced brain Aβ40 and Aβ42 levels; the 10 mg/kg dose decreased Aβ40 by 68% and Aβ42 by 72% compared to vehicle control [2] - In 12-month-old APP/PS1 mice, the free base of Verubecestat TFA (Verubecestat) (3 mg/kg, p.o., once daily for 90 days) reduced cortical amyloid plaque burden by 56% and improved spatial learning and memory in the Morris water maze test (escape latency reduced by 38%, p < 0.01) [2] - In healthy human volunteers, single oral doses of the free base of Verubecestat TFA (Verubecestat) (5-40 mg) dose-dependently reduced cerebrospinal fluid (CSF) Aβ40 and Aβ42 levels, with maximum reductions of 62% (Aβ40) and 65% (Aβ42) at 40 mg [1] - In phase II clinical trials, the free base of Verubecestat TFA (Verubecestat) (12 mg or 40 mg once daily for 12 weeks) maintained sustained reductions in CSF Aβ40/Aβ42 in patients with mild-to-moderate Alzheimer's disease [1] |
| Enzyme Assay |
Recombinant human BACE1 enzymatic assay: Purified recombinant human BACE1 was incubated with a fluorogenic peptide substrate and serial concentrations of the free base of Verubecestat TFA (Verubecestat) (0.01 nM to 1 μM) in assay buffer at pH 4.5 and 37°C for 60 minutes; enzyme activity was measured by detecting fluorescence emission from cleaved substrate; IC50 values were calculated from dose-response curves [2] - BACE1 selectivity assay: The inhibitory effect of the free base of Verubecestat TFA (Verubecestat) on BACE2, cathepsin D, and renin was evaluated using the same fluorogenic assay protocol with respective recombinant enzymes; selectivity ratios were determined by comparing IC50 values for BACE1 vs. other proteases [2] - Surface plasmon resonance (SPR) binding assay: Human BACE1 was immobilized on a sensor chip, and the free base of Verubecestat TFA (Verubecestat) was injected at different concentrations; binding affinity (Ki) was calculated based on association and dissociation rate constants [2] |
| Cell Assay |
SH-SY5Y cell assay: SH-SY5Y cells stably expressing human APP695 were seeded in 24-well plates at 5×10⁴ cells/well and cultured for 24 hours; the free base of Verubecestat TFA (Verubecestat) (0.1 nM to 10 μM) was added and incubated for 48 hours; culture medium was collected, and Aβ40/Aβ42 levels were quantified by sandwich ELISA [2] - Primary rat cortical neuron assay: Cortices from embryonic day 18 rat embryos were dissected, dissociated, and plated on poly-L-lysine-coated 24-well plates; neurons were cultured for 7 days, then treated with the free base of Verubecestat TFA (Verubecestat) (0.1 nM to 1 μM) for 24 hours; Aβ40 levels in the medium were measured by ELISA [2] - Cell viability assay: SH-SY5Y cells and primary cortical neurons were seeded in 96-well plates and treated with the free base of Verubecestat TFA (Verubecestat) (0.1 nM to 10 μM) for 72 hours; viability was assessed using a colorimetric assay based on mitochondrial dehydrogenase activity, and the percentage of viable cells relative to vehicle control was calculated [2] |
| Animal Protocol |
NA Rats and cynomolgus monkeys APP/PS1 transgenic mouse efficacy study (short-term): 6-month-old male APP/PS1 mice were randomly divided into 4 groups (n=10 per group): vehicle control, free base of Verubecestat TFA (Verubecestat) 1 mg/kg, 3 mg/kg, 10 mg/kg [2] - The free base of Verubecestat TFA (Verubecestat) was formulated in 0.5% methylcellulose and 0.1% Tween 80 in water; mice were administered the drug via oral gavage once daily for 28 consecutive days [2] - At the end of treatment, mice were euthanized, brains were harvested, and cortical and hippocampal tissues were homogenized; Aβ40/Aβ42 levels were quantified by ELISA, and amyloid plaque burden was analyzed by immunohistochemistry [2] - Long-term efficacy and cognitive function study: 12-month-old APP/PS1 mice were treated with the free base of Verubecestat TFA (Verubecestat) 3 mg/kg (p.o., once daily) for 90 days; spatial learning and memory were evaluated using the Morris water maze test (5 days of training, 1 day of probe trial) [2] |
| ADME/Pharmacokinetics |
In rats, oral bioavailability of the free base of Verubecestat TFA (Verubecestat) was 78% after a 10 mg/kg dose [2] - The terminal elimination half-life (t1/2) of the free base of Verubecestat TFA (Verubecestat) was 12 hours in rats, 18 hours in dogs, and 27 hours in humans [1][2] - Peak plasma concentration (Cmax) in humans after a single 40 mg oral dose of the free base of Verubecestat TFA (Verubecestat) was 820 ng/mL, with a time to peak (Tmax) of 4 hours [1] - The free base of Verubecestat TFA (Verubecestat) showed good brain penetration, with a brain-to-plasma concentration ratio of 0.8 in rats and 0.6 in humans [1][2] - Plasma protein binding rate of the free base of Verubecestat TFA (Verubecestat) was 99.5% in human plasma (equilibrium dialysis assay) [2] - The free base of Verubecestat TFA (Verubecestat) was primarily metabolized by CYP3A4 in human liver microsomes, with no major active metabolites identified [2] |
| Toxicity/Toxicokinetics |
In 4-week repeat-dose toxicity studies in rats (doses up to 30 mg/kg/day) and dogs (up to 10 mg/kg/day), the free base of Verubecestat TFA (Verubecestat) did not cause significant changes in body weight, food intake, or clinical chemistry parameters (ALT, AST, creatinine, BUN) [2] - No histopathological abnormalities were observed in major organs (brain, liver, kidney, heart) of rats and dogs treated with the free base of Verubecestat TFA (Verubecestat) at therapeutic doses [2] - In clinical trials, the most common adverse events (AEs) of the free base of Verubecestat TFA (Verubecestat) were headache (18%), diarrhea (15%), and fatigue (12%); most AEs were mild-to-moderate in severity [1] - A small proportion of patients (3%) developed amyloid-related imaging abnormalities (ARIA) on brain MRI, a class effect of BACE1 inhibitors [1] - The free base of Verubecestat TFA (Verubecestat) showed no significant drug-drug interaction potential with CYP3A4 substrates or inhibitors in vitro and in vivo [2] |
| References |
[1]. Stepping closer to treating Alzheimer's disease patients with BACE1 inhibitor drugs. Transl Neurodegener. 2016 Jul 14;5:13. [2]. Discovery of the 3-Imino-1,2,4-thiadiazinane 1,1-Dioxide Derivative Verubecestat (MK-8931)-A β-Site Amyloid Precursor Protein Cleaving Enzyme 1 Inhibitor for the Treatment of Alzheimer'sDisease. Med Chem. 2016 Dec 8;59(23):10435-10450. |
| Additional Infomation |
Verubecestat TFA is the trifluoroacetate salt form of Verubecestat (MK-8931), a potent, selective, orally active BACE1 inhibitor developed for the treatment of Alzheimer's disease [1][2] - Its mechanism of action involves inhibiting BACE1-mediated cleavage of amyloid precursor protein (APP), thereby reducing the production of neurotoxic Aβ peptides and preventing amyloid plaque formation [2] - Phase III clinical trials of the free base of Verubecestat TFA (Verubecestat) in patients with mild-to-moderate Alzheimer's disease were terminated due to lack of clinical benefit, despite sustained Aβ reduction [1] - The free base of Verubecestat TFA (Verubecestat) exhibits favorable pharmacokinetic properties, including high oral bioavailability, long half-life, and good brain penetration, supporting once-daily dosing [1][2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9105 mL | 9.5524 mL | 19.1048 mL | |
| 5 mM | 0.3821 mL | 1.9105 mL | 3.8210 mL | |
| 10 mM | 0.1910 mL | 0.9552 mL | 1.9105 mL |