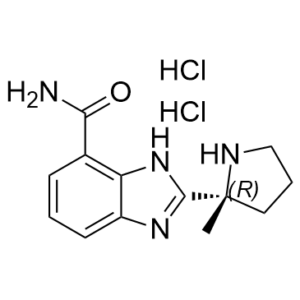
Veliparib dihydrochloride (also known as ABT888 dihydrochloride; ABT-888 dihydrochloride) is a novel and potent inhibitor of PARP1 and PARP2 [Poly (ADP-Ribose) Polymerase] with Ki of 5.2 nM and 2.9 nM in cell-free assays, respectively. It is inert against SIRT2 and exhibits chemosensitizing and antitumor properties. When combined with a range of cytotoxic agents, ABT-888 has shown excellent in vivo efficacy in a broad spectrum of preclinical tumor models. Compared to microsatellite stable (MSS) cell lines (wild-type for both genes), ABT-888 is also active on microsatellite instability (MSI) cell lines carrying mutations in both the MRE11 and RAD50 genes. At therapeutic concentrations, ABT-888 has no antiproliferative effects. Instead, it inhibits PARPs, which prevents DNA repair and increases the cytotoxicity of agents that damage DNA.
Physicochemical Properties
| Molecular Formula | C13H18CL2N4O | |
| Molecular Weight | 317.21 | |
| Exact Mass | 316.085 | |
| Elemental Analysis | C, 49.22; H, 5.72; Cl, 22.35; N, 17.66; O, 5.04 | |
| CAS # | 912445-05-7 | |
| Related CAS # | 912444-00-9; 912445-05-7 (HCl) | |
| PubChem CID | 45480520 | |
| Appearance | White to off-white solid powder | |
| LogP | 3 | |
| Hydrogen Bond Donor Count | 5 | |
| Hydrogen Bond Acceptor Count | 3 | |
| Rotatable Bond Count | 2 | |
| Heavy Atom Count | 20 | |
| Complexity | 348 | |
| Defined Atom Stereocenter Count | 1 | |
| SMILES | C(C1C=CC=C2N=C([C@@]3(NCCC3)C)NC=12)(=O)N.Cl |
|
| InChi Key | DSBSVDCHFMEYBX-FFXKMJQXSA-N | |
| InChi Code | InChI=1S/C13H16N4O.2ClH/c1-13(6-3-7-15-13)12-16-9-5-2-4-8(11(14)18)10(9)17-12;;/h2,4-5,15H,3,6-7H2,1H3,(H2,14,18)(H,16,17);2*1H/t13-;;/m1../s1 | |
| Chemical Name | 2-[(2R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide;dihydrochloride | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | PARP-2 ( Ki = 2.9 nM ); PARP-1 ( Ki = 5.2 nM ) |
| ln Vitro | Veliparib exhibits no effect on SIRT2 (>5 μM). [1] With an EC50 of 2 nM, veliparib suppresses PARP activity in C41 cells[2]. In H460 cells exposed to and not exposed to light, veliparib lowers PAR levels. DNA repair and clonogenesis in H460 cells are inhibited by PARP-1 inhibition. When veliparib is combined with radiation therapy, H460 cells exhibit increased sequestration and autophagy [3]. In H1299, DU145, and 22RV1 cells, veliparib decreases PARP activity; this suppression is correlated with p53 function. In clonogenic H1299 cells, veliparib (10 μM) reduces quarter fraction (SF) by 43%. In oxygenated H1299 cells, velibib distributes radiation efficiently. In hypoxia-irradiated cells such H1299, DU145, and 22RV1, veliparib can disconnect SF[4]. |
| ln Vivo |
Veliparib has a 56%–92% postcranial bioavailability in mice, SD spray, beagle dogs, and cynomolgus monkeys[1]. Tumor growth delay is improved in the NCI-H460 xenograft model by veliparib (25 mg/kg, ip). Veliparib can lessen tumor vascularization when combined with radiation therapy [3]. Veliparib, at dosages of 3 and 12.5 mg/kg, decreased intratumoral PAR levels in A375 and Colo829 xenograft models by more than 95%; this suppression was reversible over time [4].
Veliparib (ABT-888) potentiates temozolomide in a syngeneic melanoma model. [1] Temozolomide is a newer generation of cytotoxic alkylating agent that is currently used to treat central nervous system malignancies and melanoma. The pharmacokinetic profile of temozolomide is similar between mice and humans, and this allows studies in mice at similar exposures to those achieved in humans. This is important because preclinical models best predict clinical outcomes when plasma drug concentrations of the cytotoxic agents are similar to those seen in humans. Consequently, a dose of 50 to 62.5 mg/kg/d temozolomide was used, and this dose closely mimics human exposure at the clinically relevant dose of 200 mg/m2 (oral, q.d.×5) when measured by either area under the concentration curve (AUC) or Cmax. At this dose range, no overt toxicity (e.g., excessive weight loss, ruffled coats, dehydration, etc.) was observed in mice. [1] The B16 model, which is relatively resistant to most chemotherapeutics, is moderately sensitive to temozolomide and its sensitivity can be enhanced with PARP inhibitors. Veliparib (ABT-888), administered orally, significantly potentiated the temozolomide efficacy in a dose-dependent manner (Fig. 2A). Maximum potentiation was seen at day 19 with % T/C values (versus temozolomide) of 10 (P = 0.0003), 16 (P < 0.0001), and 23 (P < 0.0001) for the 25, 12.5, and 3.1 mg/kg/d Veliparib (ABT-888) combination groups, respectively. The combinations were well tolerated with maximum body weight loss of 11% for the 25 mg/kg/d ABT-888 and temozolomide combination compared with 7% for temozolomide and 2% for ABT-888. The mice rapidly regained weight once the dosing period ended. [1] To establish the steady-state concentration necessary for in vivo activity, Veliparib (ABT-888) was administered as a continuous infusion in combination with 50 mg/kg/d temozolomide. ABT-888 at doses of 25 to 1 mg/kg/d all significantly potentiated the temozolomide monotherapy (Fig. 2B). Maximum potentiation was seen at day 17 with % T/C values (versus temozolomide) of 13 (P < 0.0001), 12 (P < 0.0001), 16 (P < 0.0001), 39 (P = 0.0033), and 63 (not significant) for the 25, 12.5, 5, 1, and 0.3 mg/kg/d ABT-888 combination groups, respectively. A higher dose of 50 mg/kg/d ABT-888 could not be evaluated with temozolomide because this combination resulted in skin toxicity at the OMP implantation site. The 25 and 12.5 mg/kg/d ABT-888 combination treatments were equivalent in activity, thereby defining the maximally efficacious dose as 12.5 mg/kg/d in this model. Maximum weight loss for the combination groups was 1% compared with a 5% gain for temozolomide. [1] In vivo inhibition of PAR. [1] Activation of PARP in response to DNA damage results in ribosylation of various substrate proteins. To show inhibition of PARP activity in vivo, tumors from mice treated with Veliparib (ABT-888) were analyzed for levels of PAR by Western blot. B16F10 tumor-bearing mice were treated with either vehicle, temozolomide alone, or in combination with ABT-888 (Fig. 2C), similar to doses used in the efficacy study (Fig. 2A). A decrease in PAR proteins in tumors from animals treated with either ABT-888 alone or in combination with temozolomide was observed, indicating the ability of ABT-888 to inhibit PARP activity in vivo. [1] Veliparib (ABT-888) potentiates temozolomide in a syngeneic glioma model. ABT-888 also potentiates temozolomide in a 9L orthotopic rat glioma model. Maximum efficacy was seen at day 14 using magnetic resonance imaging (Fig. 3A and C). ABT-888 at 50 mg/kg/d in combination with temozolomide reduced tumor volume by 63%, which was 44% better than temozolomide alone (P < 0.005). Tumor growth inhibition with ABT-888 was dose dependent (Fig. 3A and B). The combination of 50 mg/kg/d dose of ABT-888 with temozolomide significantly prolonged animal survival versus temozolomide with a median survival of 19 and 22 days, respectively (P < 0.0132, log-rank test; Fig. 3B). ABT-888 as a single agent at 50 mg/kg/d was not efficacious in this model (data not shown). The combinations were well tolerated with maximum weight losses of only 9% for the combination compared with 8% for temozolomide. Veliparib (ABT-888) potentiates platinum agents. [1] The ability of ABT-888 to potentiate the efficacy of platinum-based agents was investigated in the MX-1 breast carcinoma xenograft model. This line was derived from a 29-year-old female with a poorly differentiated mammary carcinoma. Internal sequencing efforts at Abbott have determined that MX-1 has BRCA1 deletions. A novel BRCA1 variant (BRCA1 33636delGAAA) was detected that would result in a frameshift mutation predicted to introduce a chain terminator and truncate the protein at residue 999. Two previously described nonsynomous single-nucleotide polymorphisms were also detected in BRCA2 (BRCA2 16864A>C, Asn289His, and BRCA2 221847A>G, Asn991Asp), both of which have been described in Chinese Breast Cancer families. Veliparib (ABT-888) induced a pronounced potentiation of cisplatin activity (Fig. 4A). At day 68 when all mice were still present in each combination group, no significant differences were noted between the 5, 25, and 50 mg/kg/d combinations with cisplatin. However, at the end of the trial, ABT-888 at 5, 25, and 50 mg/kg/d in combination with cisplatin showed an increase in cures (8/9, 8/9, and 6/9 animals, respectively), whereas the cisplatin monotherapy had only 3/9 cures (no measurable tumors at end of the trial). Both the 5 and 25 mg/kg/d ABT-888 plus cisplatin were significantly different than cisplatin alone (P = 0.049, Fisher's exact test), whereas the 50 mg/kg/d ABT-888 was not significantly different from 5 and 25 mg/kg/d treatment combinations. The vehicle and ABT-888 monotherapy groups had no cures. This dose-response study showed that maximal potentiation was reached at 5 mg/kg/d ABT-888. Potentiation of platinum agents was confirmed in a second MX-1 study using carboplatin. Carboplatin is a second-generation platinum, less toxic anticancer drug and is currently the standard of care for treating lung, ovarian, and head and neck cancers. ABT-888 administered at 25 mg/kg/d via OMPs caused a pronounced potentiation of carboplatin at 10 and 15 mg/kg/d as reflected by tumor volumes (Fig. 4B). Compared with carboplatin treatment groups at day 38, the %T/C values were 34 (P = 0.011) and 18 (P < 0.0001) for the carboplatin 15 and 10 mg/kg/d combinations with ABT-888, respectively. The 10 mg/kg/d carboplatin/ABT-888 combination regressed tumor volumes from day 26, whereas carboplatin monotherapy had only a modest tumor inhibition. Because Veliparib (ABT-888) showed a pronounced potentiation of carboplatin at 10 mg/kg/d, a separate study was undertaken to determine the dose-response relationship of ABT-888 at a fixed carboplatin dose. ABT-888 potentiated the activity of 10 mg/kg/d carboplatin (q4d×3) with %T/C values (versus carboplatin) at day 42 of 9 (P < 0.0005), 22 (P = 0.0014), and 42 (P = 0.012) for the 50, 25, and 12.5 mg/kg/d combinations of ABT-888 and carboplatin (data not shown). The 5 and 1 mg/kg/d doses of ABT-888 did not potentiate carboplatin. Veliparib (ABT-888) potentiates cyclophosphamide. [1] In the MX-1 model, ABT-888 administered at 25 mg/kg/d via OMPs not only potentiated cyclophosphamide at 12.5 mg/kg/d on days 20, 24, and 27 schedule (Fig. 4C) but also caused tumor regression, whereas the cyclophosphamide monotherapy only slightly delayed tumor growth. The %T/C values of this combination (versus cyclophosphamide) at day 38 was 35 (P = 0.0011). Cyclophosphamide was not effective at 5 mg/kg/d and ABT-888 did not potentiate the cytotoxic agent at this dose. In a separate confirmatory study, ABT-888 at 25 mg/kg/d enhanced the efficacy of cyclophosphamide (12.5 mg/kg/d, q4d×3) but potentiation was not shown at 12.5 mg/kg/d of ABT-888. The %T/C values of combinations (versus carboplatin) at day 42 were 53 (P = 0.018), 91 (not significant), and 100 (not significant) for the 25, 12.5, and 5 mg/kg/d doses of ABT-888 combinations, respectively (data not shown). The ability of Veliparib (ABT-888) to potentiate the efficacy of cyclophosphamide was also evaluated in the DOHH-2 B-cell lymphoma flank xenograft model. DOHH-2 is a lymphoma line with the t(14;18) translocation that results in expression of high levels of Bcl-2 and is sensitive to cyclophosphamide. However, ABT-888 did not potentiate cyclophosphamide using an array of cytotoxic schedules (q.d.×1, q.d.×4, q4d×2, and q4d×3) and dosing schemes (data not shown), although cyclophosphamide showed single-agent activity. These data indicate that ABT-888 is not a potentiator of cyclophosphamide in the DOHH-2 model. Veliparib (ABT-888)potentiates radiation. [1] HCT-116 is a human colon cancer line that has been very well characterized for radiation sensitivity, including growth delay, cell cycle arrest, and apoptosis. ABT-888 administered via OMPs at 25 mg/kg/d potentiated fractionated radiation (2 Gy/d × 10 days) with a median survival time of 36 days compared with 23 days (P < 0.036, log-rank test) from radiation alone (Fig. 5). Although ABT-888 did not enhance median survival (34 days) at 12.5 mg/kg/d (P = 0.06), this treatment group did have one cure (no palpable tumor) when the study was terminated on day 65. ABT-888 was also tested at 5 and 1 mg/kg/d in combination with radiation but these groups were not significantly different than radiation alone. Overall, ABT-888 showed a dose response in combination with radiation (P = 0.0165, log-rank trend). |
| Enzyme Assay |
In PARP assays, 50 mM Tris (pH 8.0), 1 mM DTT, 1.5 μM [3H]NAD+ (1.6 μCi/mmol), 200 nM biotinylated histone H1, 200 nM slDNA, and 1 nM PARP-1 or 4 nM PARP-2 enzyme are added to a buffer. After 1.5 mM benzamide is added to stop the reaction, the reaction is moved to streptavidin Flash plates and counted using a TopCount microplate scintillation counter.
Using a commercial assay kit, PARP1 enzyme activity is measured; however, instead of using the PARP1 protein that comes with the kit, cell lysates containing either the wild-type PARP1 or the PARP Y907 mutant are used. Each reaction receives 500 ng of the entire lysate. Veliparib (ABT-888), a PARP inhibitor, has a dosage range of 0.01 to 1,000 μM. A plate reader is used to measure the PARP enzyme activity of both wild-type and mutant samples following their incubation with the substrate[2]. In vitro PARP and SIRT assays. PARP assays were conducted in a buffer containing 50 mmol/L Tris (pH 8.0), 1 mmol/L DTT, 1.5 μmol/L [3H]NAD+ (1.6 μCi/mmol), 200 nmol/L biotinylated histone H1, 200 nmol/L slDNA, and 1 nmol/L PARP-1 or 4 nmol/L PARP-2 enzyme. Reactions were terminated with 1.5 mmol/L benzamide, transferred to streptavidin Flash plates, and counted using a TopCount microplate scintillation counter.[1] Nicotinamide [2,5′,8-3H]adenine dinucleotide and streptavidin SPA beads were purchased from xxx Biosciences. Recombinant human PARP purified from Escherichia coli and 6-Biotin-17-NAD+ were purchased from xxx. NAD+, histone, aminobenzamide, 3-aminobenzamide, and calf thymus DNA (dcDNA) were from xxx. Stem loop oligonucleotide (slDNA) CACAAGTGTTGCATTCCTC-TCTGAAGTTAAGACCTATGCAGAGAGGAATGCAACACTTGTG, containing MCAT sequence (italics), was obtained from Qiagen. The oligonucleotides were dissolved to 1 mmol/L in annealing buffer containing 10 mmol/L Tris-HCl (pH 7.5), 1 mmol/L EDTA, and 50 mmol/L NaCl, incubated for 5 min at 95°C, and followed by annealing at 45°C for 45 min. Histone H1 (95% electrophoretically pure) was purchased from yyy. Biotinylated histone H1 was prepared by treating the protein with Sulfo-NHS-LC-Biotin. SIRT2 assays were conducted as described previously. |
| Cell Assay |
Cellular PARP Assay[2] C41 cells were treated with test compound for 30 min in a 96-well plate. PARP was activated by damaging DNA with 1 mM H2O2 for 10 min. Cells were washed with ice-cold PBS once and fixed with prechilled methanol/acetone (7:3) at −20 °C for 10 min. After air-drying, plates were rehydrated with PBS and blocked using 5% nonfat dry milk in PBS-tween (0.05%) (blocking solution) for 30 min at room temperature. Cells were incubated with anti-PAR antibody 10H (1:50) in blocking solution at room temperature for 60 min followed by washing with PBS-Tween20 5 times and incubation with goat antimouse fluorescein 5(6)-isothiocyanate (FITC)-coupled antibody (1:50) and 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) in blocking solution at room temperature for 60 min. After washing with PBS-Tween20 5 times, analysis was performed using an fmax Fluorescence Microplate Reader set at the excitation and emission wavelength for FITC or the excitation and emission wavelength for DAPI. PARP activity (FITC signal) was normalized with cell numbers (DAPI). |
| Animal Protocol | In order to conduct syngeneic studies on B16F10, a mixture of 6×104 cells and 50% Matrigel is injected subcutaneously (20 g) into the flank of 6- to 8-week-old female C57BL/6 mice. In order to conduct cisplatin efficacy studies, fragments (20–30 mm3) of human tumors taken from spontaneously growing tumors in nude mouse hosts are trocar-implanted in female nude mice. In the studies involving carboplatin and MX-1 cyclophosphamide, 200 μL of a 1:10 dilution of tumor brei in 45% Matrigel and 45% Spinner MEM is administered to female scid mice as an injection. Tumors are allowed to grow to the specified size in these well-established tumor studies, after which they are randomized to therapy groups. Male scid mice are given a s.c. injection of 1×106 cells mixed with 50% Matrigel to be used in DOHH-2 xenograft studies. Veliparib is administered orally or continuously via s.c. insertion of a 14-day Alzet OMP model 2002 in a pH 4.0-adjusted 0.9% NaCl solution. Doses of Veliparib are determined based on the OMP's daily delivery rate of 12 μL. The formulation of temozolomide, cisplatin, carboplatin, and cyclophosphamide is done in accordance with the advice of the manufacturers. |
| ADME/Pharmacokinetics |
Pharmacokinetics Results [5] PK parameters in the group with dosages greater than or equal to 200 mg BID (high veliparib, n=18) and those less than 200 mg BID (low veliparib, n=7) were compared, utilizing an unpaired, 2 sided t-test. PK studies were performed on 25 of 31 patients with complete samples. We noted a positive correlation between the AUC/mg of PLD (area under the curve on a plot of drug concentration in blood plasma vs time) and veliparib dose, p=0.001, and a negative correlation between PLD clearance (CL), p=0.001 and veliparib dose. When analyzed as low and high veliparib groups, the mean (SD) half-life (hrs) was significantly lower in the low veliparib when compared to the high veliparib group, 83.2 (33.2) vs 108.6 (24.88) (p =0.042), and the mean PLD clearance (mL/h) was greater in the low veliparib versus the high veliparib group 35.9 (15.9) vs 14.2 (4.3), P<.0001. Similarly, the PLD AUC/mg dose (mg x h/L) was significantly lower in the low vs high veliparib groups, 35.0 (14.7)vs 74.9 (18.3), P<0.0001. Thirteen patients had evaluable veliparib PK from the expanded cohort (see supplement). The half-life was slightly shorter at 4.1 h on day 1 compared to 5.3 h on day 8. The day 8 apparent clearance was 15.4 L/h and apparent volume of distribution was 121 L with both parameters calculated using day 8 AUC0–12. Dose-normalizing exposure to accommodate dose reductions between days 1 and 8 resulted in statistical significance of day 1 Cmax vs predicted day 8 Cmax (Fig. 3A, p-value=0.0022) and day 1 AUC0−∞ vs day 8 AUC0–12 (Fig 3B, p-value 0.0151). Observed accumulation ratios were lower than the expected 1.2 accumulation ratio that was calculated with the observed half-life and 12 h dosing interval. The observed accumulation ratio for Cmax was 0.865, AUC0–12 was 0.816, and AUC0–12/ AUC0−∞ was 0.666. |
| Toxicity/Toxicokinetics | The majority of patients (39%) were treated at 200 mg of veliparib BID (final dose reached after de-escalation) and 70% were treated with 40 mg/m2 PLD. Dose de-escalation of veliparib or PLD occurred due to adverse events in 20 (49%) patients in stratum B and 2 (67%) in stratum A, including patients with a DLT. Table 3 shows adverse events. Although grade 1 and 2 toxicities were common, only 10% patients experienced a grade 3 or 4 toxicity from this therapy regimen. The most common grade 3 and 4 adverse events included: anemia, 4 (10%); neutropenia, 4 (10%); hand-foot syndrome (HFS), 4 (10%); nausea, 2 (5%); vomiting, 2 (5%); neuropathy, 2 (5%); and transaminitis, 2 (5%). Therapy was discontinued for 5 (11%) patients due to toxicity. In stratum A, a patient developed grade 4 pericardial tamponade at 200 mg veliparib BID and 40 mg/m2 PLD (after 1 cycle). Cytology from the pericardial fluid was positive for malignant cells. In stratum B therapy was discontinued due unresolved thrombocytopenia at 50 mg veliparib BID and 22.5 mg/m2 PLD (after 24 cycles), grade 3 HFS at 50 mg veliparib BID and 22.5 mg/m2 PLD (after 32 cycles), a decreased left ventricular ejection fraction (52%, vs 72% LVEF prior to treatment) at 200 mg veliparib BID and 40 mg/m2 PLD (after 5 cycles), and unresolved leukopenia at 300 mg veliparib BID and 40 mg/m2 PLD (after 2 cycles).[5] |
| References |
[1]. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007 May 1;13(9):2728-37. [2]. Discovery of the Poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the treatment of cancer. J Med Chem. 2009 Jan 22;52(2):514-23. [3]. Inhibition of poly(ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin Cancer Res. 2007 May 15;13(10):3033-42. [4]. Preclinical Modeling of a Phase 0 Clinical Trial: Qualification of a Pharmacodynamic Assay of Poly (ADP-Ribose) Polymerase in Tumor Biopsies of Mouse Xenografts. Clin Cancer Res. Author manuscript; available in PMC 2009 Nov 1. [5]. Phase I and Pharmacokinetic Study of Veliparib, a PARP Inhibitor, and Pegylated Liposomal Doxorubicin (PLD) in Recurrent Gynecologic Cancer and Triple Negative Breast Cancer with Long-Term Follow-Up. Cancer Chemother Pharmacol. 2020 Feb 13;85(4):741–751 |
| Additional Infomation |
Veliparib is a benzimidazole substituted with a carbamoyl group at C-4 and a (2R)-2-methylpyrrolidin-2-yl moiety at C-2. It is a potent, orally bioavailable PARP inhibitor. It has a role as an EC 2.4.2.30 (NAD(+) ADP-ribosyltransferase) inhibitor.
< Veliparib is a poly(ADP-ribose) polymerase (PARP) -1 and -2 inhibitor with chemosensitizing and antitumor activities. With no antiproliferative effects as a single agent at therapeutic concentrations, ABT-888 inhibits PARPs, thereby inhibiting DNA repair and potentiating the cytotoxicity of DNA-damaging agents. PARP nuclear enzymes are activated by DNA single or double strand breaks, resulting in the poly(ADP-ribosyl)ation of other nuclear DNA binding proteins involved in DNA repair; poly(ADP-ribosyl)ation contributes to efficient DNA repair and to survival of proliferating cells exposed to mild genotoxic stresses as induced by as oxidants, alkylating agents or ionizing radiation. Drug Indication Treatment of high-grade glioma; Treatment of fallopian tube cancer , Treatment of ovarian cancer , Treatment of peritoneal carcinoma;Treatment of lung carcinoma (small cell and non-small cell carcinoma) Veliparib is a benzimidazole substituted with a carbamoyl group at C-4 and a (2R)-2-methylpyrrolidin-2-yl moiety at C-2. It is a potent, orally bioavailable PARP inhibitor. It has a role as an EC 2.4.2.30 (NAD(+) ADP-ribosyltransferase) inhibitor. Veliparib is a poly(ADP-ribose) polymerase (PARP) -1 and -2 inhibitor with chemosensitizing and antitumor activities. With no antiproliferative effects as a single agent at therapeutic concentrations, ABT-888 inhibits PARPs, thereby inhibiting DNA repair and potentiating the cytotoxicity of DNA-damaging agents. PARP nuclear enzymes are activated by DNA single or double strand breaks, resulting in the poly(ADP-ribosyl)ation of other nuclear DNA binding proteins involved in DNA repair; poly(ADP-ribosyl)ation contributes to efficient DNA repair and to survival of proliferating cells exposed to mild genotoxic stresses as induced by as oxidants, alkylating agents or ionizing radiation. Drug Indication Treatment of high-grade glioma Treatment of fallopian tube cancer , Treatment of ovarian cancer , Treatment of peritoneal carcinoma Treatment of lung carcinoma (small cell and non-small cell carcinoma) Treatment of breast cancer. Purpose: To evaluate the preclinical pharmacokinetics and antitumor efficacy of a novel orally bioavailable poly(ADP-ribose) polymerase (PARP) inhibitor, ABT-888. Experimental design: In vitro potency was determined in a PARP-1 and PARP-2 enzyme assay. In vivo efficacy was evaluated in syngeneic and xenograft models in combination with temozolomide, platinums, cyclophosphamide, and ionizing radiation. Results: ABT-888 is a potent inhibitor of both PARP-1 and PARP-2 with K(i)s of 5.2 and 2.9 nmol/L, respectively. The compound has good oral bioavailability and crosses the blood-brain barrier. ABT-888 strongly potentiated temozolomide in the B16F10 s.c. murine melanoma model. PARP inhibition dramatically increased the efficacy of temozolomide at ABT-888 doses as low as 3.1 mg/kg/d and a maximal efficacy achieved at 25 mg/kg/d. In the 9L orthotopic rat glioma model, temozolomide alone exhibited minimal efficacy, whereas ABT-888, when combined with temozolomide, significantly slowed tumor progression. In the MX-1 breast xenograft model (BRCA1 deletion and BRCA2 mutation), ABT-888 potentiated cisplatin, carboplatin, and cyclophosphamide, causing regression of established tumors, whereas with comparable doses of cytotoxic agents alone, only modest tumor inhibition was exhibited. Finally, ABT-888 potentiated radiation (2 Gy/d x 10) in an HCT-116 colon carcinoma model. In each model, ABT-888 did not display single-agent activity. Conclusions: ABT-888 is a potent inhibitor of PARP, has good oral bioavailability, can cross the blood-brain barrier, and potentiates temozolomide, platinums, cyclophosphamide, and radiation in syngeneic and xenograft tumor models. This broad spectrum of chemopotentiation and radiopotentiation makes this compound an attractive candidate for clinical evaluation. [1] View MoreTo evaluate the preclinical pharmacokinetics and antitumor efficacy of a novel orally bioavailable poly(ADP-ribose) polymerase (PARP) inhibitor, ABT-888. Experimental design: In vitro potency was determined in a PARP-1 and PARP-2 enzyme assay. In vivo efficacy was evaluated in syngeneic and xenograft models in combination with temozolomide, platinums, cyclophosphamide, and ionizing radiation. Results: ABT-888 is a potent inhibitor of both PARP-1 and PARP-2 with K(i)s of 5.2 and 2.9 nmol/L, respectively. The compound has good oral bioavailability and crosses the blood-brain barrier. ABT-888 strongly potentiated temozolomide in the B16F10 s.c. murine melanoma model. PARP inhibition dramatically increased the efficacy of temozolomide at ABT-888 doses as low as 3.1 mg/kg/d and a maximal efficacy achieved at 25 mg/kg/d. In the 9L orthotopic rat glioma model, temozolomide alone exhibited minimal efficacy, whereas ABT-888, when combined with temozolomide, significantly slowed tumor progression. In the MX-1 breast xenograft model (BRCA1 deletion and BRCA2 mutation), ABT-888 potentiated cisplatin, carboplatin, and cyclophosphamide, causing regression of established tumors, whereas with comparable doses of cytotoxic agents alone, only modest tumor inhibition was exhibited. Finally, ABT-888 potentiated radiation (2 Gy/d x 10) in an HCT-116 colon carcinoma model. In each model, ABT-888 did not display single-agent activity. Conclusions: ABT-888 is a potent inhibitor of PARP, has good oral bioavailability, can cross the blood-brain barrier, and potentiates temozolomide, platinums, cyclophosphamide, and radiation in syngeneic and xenograft tumor models. This broad spectrum of chemopotentiation and radiopotentiation makes this compound an attractive candidate for clinical evaluation.[1] Poly (ADP-ribose) polymerase (PARP) inhibitors have emerged as promising therapeutics for many diseases, including cancer, in clinical trials. One PARP inhibitor, olaparib (Lynparza, AstraZeneca), was recently approved by the FDA to treat ovarian cancer with mutations in BRCA genes. BRCA1 and BRCA2 have essential roles in repairing DNA double-strand breaks, and a deficiency of BRCA proteins sensitizes cancer cells to PARP inhibition. Here we show that the receptor tyrosine kinase c-Met associates with and phosphorylates PARP1 at Tyr907 (PARP1 pTyr907 or pY907). PARP1 pY907 increases PARP1 enzymatic activity and reduces binding to a PARP inhibitor, thereby rendering cancer cells resistant to PARP inhibition. The combination of c-Met and PARP1 inhibitors synergized to suppress the growth of breast cancer cells in vitro and xenograft tumor models, and we observed similar synergistic effects in a lung cancer xenograft tumor model. These results suggest that the abundance of PARP1 pY907 may predict tumor resistance to PARP inhibitors, and that treatment with a combination of c-Met and PARP inhibitors may benefit patients whose tumors show high c-Met expression and who do not respond to PARP inhibition alone.[2] Early studies with first-generation poly (ADP-ribose) polymerase (PARP) inhibitors have already indicated some therapeutic potential for sulfur mustard (SM) injuries. The available novel and more potential PARP inhibitors, which are undergoing clinical trials as drugs for cancer treatment, bring it back to the centre of interest. However, the role of PARP-1 in SM-induced injury is not fully understood. In this study, we selected a high potent specific PARP inhibitor ABT-888 as an example to investigate the effect of PARP inhibitor in SM injury. The results showed that in both the mouse ear vesicant model (MEVM) and HaCaT cell model, PARP inhibitor ABT-888 can reduce cell damage induced by severe SM injury. ABT-888 significantly reduced SM induced edema and epidermal necrosis in MEVM. In the HaCaT cell model, ABT-888 can reduce SM-induced NAD(+)/ATP depletion and apoptosis/necrosis. Then, we studied the mechanism of PARP-1 in SM injury by knockdown of PARP-1 in HaCaT cells. Knockdown of PARP-1 protected cell viability and downregulated the apoptosis checkpoints, including p-JNK, p-p53, Caspase 9, Caspase 8, c-PARP and Caspase 3 following SM-induced injury. Furthermore, the activation of AKT can inhibit autophagy via the regulation of mTOR. Our results showed that SM exposure could significantly inhibit the activation of Akt/mTOR pathway. Knockdown of PARP-1 reversed the SM-induced suppression of the Akt/mTOR pathway. In summary, the results of our study indicated that the protective effects of downregulation of PARP-1 in SM injury may be due to the regulation of apoptosis, necrosis, energy crisis and autophagy. However, it should be noticed that PARP inhibitor ABT-888 further enhanced the phosphorylation of H2AX (S139) after SM exposure, which indicated that we should be very careful in the application of PARP inhibitors in SM injury treatment because of the enhancement of DNA damage.[3] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: 100 mg/mL (315.25 mM) in PBS (add these co-solvents sequentially from left to right, and one by one), clear solution; with sonication. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.1525 mL | 15.7624 mL | 31.5249 mL | |
| 5 mM | 0.6305 mL | 3.1525 mL | 6.3050 mL | |
| 10 mM | 0.3152 mL | 1.5762 mL | 3.1525 mL |