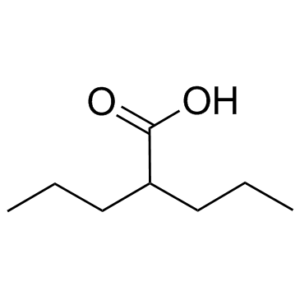
Valproic acid is an HDAC (Histone deacetylase) inhibitor used in the treatment of epilepsy, bipolar disorder and prevention of migraine headaches. Valproic Acid is a fatty acid with anticonvulsant properties by selectively inducing proteasomal degradation of HDAC2. It may act by increasing gamma-aminobutyric acid levels in the brain or by altering the properties of voltage dependent sodium channels. It is also under investigation for treatment of HIV and various cancers.
Physicochemical Properties
| Molecular Formula | C8H16O2 | |
| Molecular Weight | 144.21144 | |
| Exact Mass | 144.115 | |
| Elemental Analysis | C, 66.63; H, 11.18; O, 22.19 | |
| CAS # | 99-66-1 | |
| Related CAS # | Valproic acid sodium;1069-66-5;Valproic acid-d4;87745-17-3;Valproic acid-d6;87745-18-4;Valproic acid-d15;362049-65-8;Valproic acid (sodium)(2:1);76584-70-8;Valproic acid-d4 sodium;Valproic acid-d4-1;345909-03-7 | |
| PubChem CID | 3121 | |
| Appearance | Colorless to light yellow liquid | |
| LogP | 2.8 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 2 | |
| Rotatable Bond Count | 5 | |
| Heavy Atom Count | 10 | |
| Complexity | 93.4 | |
| Defined Atom Stereocenter Count | 0 | |
| InChi Key | NIJJYAXOARWZEE-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C8H16O2/c1-3-5-7(6-4-2)8(9)10/h7H,3-6H2,1-2H3,(H,9,10) | |
| Chemical Name | 2-propylpentanoic acid | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | HDAC1 ( IC50 = 400 μM ); HDAC1 ( IC50 = 0.5-2 mM ); HDAC2; Autophagy; Mitophagy |
| ln Vitro | In a dose- and time-dependent way, valproic acid (VPA) (0–15 mM; 24 and 72 h) suppresses the proliferation of Hela cells[1]. The activity of nuclear, cytosolic, and total HDACs is markedly reduced by valproic acid (10 mM; 24 h)[1]. The percentage of sub-G1 cells in HeLa cells rises when valproic acid (0–15 mM; 24 h) generates a G1/M phase arrest at 10 mM and a G1 phase arrest at 1-3 mM. Necrosis, apoptosis, and the release of lactate dehydrogenase (LDH) are additional effects of valproic acid[1]. Lithium works in concert with valproic acid (0–20 mM; 24 h) to stimulate Tcf/Lef-dependent transcription[2]. Neuro2A cells' β-catenin levels are elevated by valproic acid (0–5 mM; 0–18 h)[2]. Hepatocyte AMPK and ACC phosphorylation is stimulated by valproic acid (0–2 mM; 0–24 h)[5]. For two days, valproic acid (0 -10 mM) inhibits the generation of NE tumor markers in SCLC cells while inducing Notch1 signaling and morphologic differentiation[6]. |
| ln Vivo | In mice transplanted with Kasumi-1 cells, valproic acid (VPA) (500 mg/kg; ip; daily for 12 days) suppresses tumor angiogenesis[3]. Rats with valproic acid (350 mg/kg; ip; once) exhibit improved social behavior [4]. In obese mice, valproic acid (0.26% w/v; po via drinking water; 14 days) reduces blood glucose, hepatic fat formation, and liver mass without causing hepatotoxicity[5]. |
| Enzyme Assay | Valproic acid is widely used to treat epilepsy and bipolar disorder and is also a potent teratogen, but its mechanisms of action in any of these settings are unknown. We report that valproic acid activates Wntdependent gene expression, similar to lithium, the mainstay of therapy for bipolar disorder. Valproic acid, however, acts through a distinct pathway that involves direct inhibition of histone deacetylase (IC(50) for HDAC1 = 0.4 mm). At therapeutic levels, valproic acid mimics the histone deacetylase inhibitor trichostatin A, causing hyperacetylation of histones in cultured cells. Valproic acid, like trichostatin A, also activates transcription from diverse exogenous and endogenous promoters. Furthermore, valproic acid and trichostatin A have remarkably similar teratogenic effects in vertebrate embryos, while non-teratogenic analogues of valproic acid do not inhibit histone deacetylase and do not activate transcription. Based on these observations, we propose that inhibition of histone deacetylase provides a mechanism for valproic acid-induced birth defects and could also explain the efficacy of valproic acid in the treatment of bipolar disorder.[2] |
| Cell Assay |
Cell Viability Assay[1] Cell Types: HeLa cells Tested Concentrations: 0, 1, 3, 5, 10 and 15 mM Incubation Duration: 24 and 72 h Experimental Results: HeLa cell growth was dose- and time-dependently diminished with an IC50 of ~10 and 4 mM at 24 and 72 h. Western Blot Analysis[1][2][5] Cell Types: HeLa cells, Neuro2A cells or primary mouse hepatocytes Tested Concentrations: 10mM (HeLa); 0, 2, and 5mM (Neuro2A); 0.2, 0.4, 0.8, 1.2 and 2mM (hepatocytes) Incubation Duration: 10mM (HeLa ); 0, 2, and 5 mM (Neuro2A); 0.2, 0.4, 0.8, 1.2 and 2 Mm (hepatocytes) Experimental Results: Increased the form of acetylated histone 3. decreased PARP, induced cleavage PARP, and downregulated Bcl-2. Increased β-catenin levels. Increased the phosphorylation of AMPK and ACC. Cell Cycle Analysis[1] Cell Types: HeLa cells Tested Concentrations: 0, 1, 3, 5, 10 and 15 mM Incubation Duration: 24 h Experimental Results: Induced a G1 phase arrest at 1–3 mM, Dramatically induced a G2/M phase arrest at 10 mM, and increased the percentage of sub-G1 cells in HeLa cells in a dose-dependent manner at 24 |
| Animal Protocol |
Animal/Disease Models: Female BALB/c nude mice, Kasumi-1 tumor model[3] Doses: 500 mg/kg Route of Administration: intraperitoneal (ip)injection, daily for 12 days Experimental Results: Inhibited tumor growth and tumor angiogenesis. Inhibited the mRNA and protein expression of VEGF, VEGFR2 and bFGF. Inhibited HDAC activity and increased acetylation of histone H3. Enhanced the accumulation of hyperacetylated histone H3 on VEGF promoters. Animal/Disease Models: Timed-pregnant Long Evans rats[4] Doses: 350 mg/kg Route of Administration: intraperitoneal (ip)injection, once Experimental Results: Demonstrated more social investigation and play fighting than control animals. Animal/Disease Models: Obese phenotype of ob/ob mice[5] Doses: 0.26% (w/v) Route of Administration: Oral via drinking water, 14 days Experimental Results: Revealed a marked reduction in the accumulation of fats in the liver as compared with the untreated mice, Dramatically diminished liver mass to body mass, diminished serum triglyceride concentrations, and did not induce hepatotoxicity. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion The intravenous and oral forms of valproic acid are expected to produce the same AUC, Cmax, and Cmin at steady-state. The oral delayed-release tablet formulation has a Tmax of 4 hours. Differences in absorption rate are expected from other formulations but are not considered to be clinically important in the context of chronic therapy beyond impacting frequency of dosing. Differences in absorption may create earlier Tmax or higher Cmax values on initiation of therapy and may be affected differently by meals. The extended release tablet formulation had Tmax increase from 4 hours to 8 hours when taken with food. In comparison, the sprinkle capsule formulation had Tmax increase from 3.3 hours to 4.8 hours. Bioavailability is reported to be approximately 90% with all oral formulations with enteric-coated forms possibly reaching 100%. Most drug is eliminated through hepatic metabolism, about 30-50%. The other major contributing pathway is mitochondrial β-oxidation, about 40%. Other oxidative pathways make up an additional 15-20%. Less than 3% is excreted unchanged in the urine. 11 L/1.73m2. 0.56 L/hr/m2 Pediatric patients between 3 months and 10 years of age have 50% higher clearances by weight. Pediatric patients 10 years of age or older approximate adult values. Valproic acid and its salt, sodium valproate, are excreted into human milk in low concentrations. Milk concentrations up to 15% of the corresponding level in the mother's serum have been measured. In two infants, serum levels of valporate were 1.5% and 6.0% of maternal values. Placenta transfer study in non-human primate (NHP) is one of the crucial components in the assessment of developmental toxicity because of the similarity between NHP and humans. To establish the method to determine placenta transfer in non-human primate, toxicokinetics of valproic acid (VPA), a drug used to treat epilepsy in pregnant women, were determined in pregnant cynomolgus monkeys. After mating, pregnancy-proven females were daily administered with VPA at dose levels of 0, 20, 60 and 180 mg/kg by oral route during the organogenesis period from gestation day (GD) 20 to 50. Concentrations of VPA and its metabolite, 4-ene-VPA, in maternal plasma on GDs 20 and 50, and concentrations of VPA and 4-ene-VPA in placenta, amniotic fluid and fetus on GD 50 were analyzed using LC/MS/MS. Following single oral administration of VPA to pregnant monkeys, concentrations of VPA and 4-ene-VPA were generally quantifiable in the plasma from all treatment groups up to 4-24 hours post-dose, demonstrating that VPA was absorbed and the monkeys were systemically exposed to VPA and 4-ene-VPA. After repeated administration of VPA to the monkeys, VPA was detected in amniotic fluid, placenta and fetus from all treatment groups, demonstrating that VPA was transferred via placenta and the fetus was exposed to VPA, and the exposures were increased with increasing dose. Concentrations of 4-ene-VPA in amniotic fluid and fetus were below the limit of quantification, but small amount of 4-ene-VPA was detected in placenta. In conclusion, pregnant monkeys were exposed to VPA and 4-ene-VPA after oral administration of VPA at dose levels of 20, 60 and 180 mg/kg during the organogenesis period. VPA was transferred via placenta and the fetus was exposed to VPA with dose-dependent exposure. The metabolite, 4-ene VPA, was not detected in both amniotic fluid and fetus, but small amount of 4-ene-VPA was detected in placenta. These results demonstrated that proper procedures to investigate placenta transfer in NHP, such as mating and diagnosis of pregnancy via examining gestational sac with ultrasonography, collection of amniotic fluid, placenta and fetus after Caesarean section followed by adequate bioanalysis and toxicokinetic analysis, were established in this study using cynomolugus monkeys. Valproic acid - rapid absorption from GI tract; slight delay when taken with food. Protein binding is high (90 to 95%) at serum concentrations up to 50 ug/mL. As the concentration increases from 50 to 100 ug/mL, the percentage bound decreases to 80 to 85% and the free fraction becomes progressively larger, thus increasing the concentration gradient into the brain. Valproate is distributed into breast milk. Concentrations in breast milk have been reported to be 1 to 10% of the total maternal serum concentration. /Valproate/ For more Absorption, Distribution and Excretion (Complete) data for VALPROIC ACID (8 total), please visit the HSDB record page. Metabolism / Metabolites Most drug is metabolized to glucuronide conjugates (30-50%) of the parent drug or of metabolites. Another large portion is metabolized through mitochondrial β-oxidation (40%). The remainder of metabolism (15-20%) occurs through oxidation, hydroxylation, and dehydrogenation at the ω, ω1, and ω2 positions resulting in the formation of hydroxyls, ketones, carboxyls, a lactone metabolite, double bonds, and combinations. The aim of this study was to investigate the relationship between hepatotoxicity, levels of glucuronide conjugates of valproic acid (VPA), and the toxic metabolites of VPA (4-ene VPA and 2,4-diene VPA). /The study/ also examined whether hepatotoxicity could be predicted by the urinary excretion levels of VPA and its toxic metabolites. VPA was administrated orally in rats in amounts ranging from 20 mg/kg to 500 mg/kg. Free and total (free plus glucuronide conjugated) VPA, 4-ene VPA, and 2,4-diene VPA were quantified in urine and liver using gas chromatography-mass spectrometry. Serum levels of aspartate aminotransferase, alanine aminotransferase, and alpha-glutathione S-transferase (alpha-GST) were also determined to measure the level of hepatotoxicity. The serum alpha-GST level increased slightly at the 20 mg/kg dose, and substantially increased at the 100 and 500 mg/kg dose; aspartate aminotransferase and alanine aminotransferase levels did not change with the administration of increasing doses of VPA. The liver concentration of free 4-ene VPA and the urinary excretion of total 4-ene VPA were the only measures that correlated with the increase in the serum alpha-GST level (p < 0.094 and p < 0.023 respectively). From these results, /it is concluded/ that hepatotoxicity of VPA correlates with liver concentration of 4-ene VPA and can be predicted by the urinary excretion of total 4-ene VPA. BACKGROUND AND OBJECTIVE: Sodium valproate is a widely prescribed broad-spectrum antiepileptic drug. It shows high inter-individual variability in pharmacokinetics and pharmacodynamics and has a narrow therapeutic range. /This study/ evaluated the effects of polymorphic uridine diphosphate glucuronosyltransferase (UGT)1A6 (541A>G, 552A>C) metabolizing enzyme on the pharmacokinetics of sodium valproate in the patients with epilepsy who showed toxicity to therapy. METHODS: Genotype analysis of the patients was made with polymerase chain-restriction fragment length polymorphism (RFLP) with sequencing. Plasma drug concentrations were measured with reversed phase high-performance liquid chromatography (HPLC) and concentration-time data were analyzed by using a non-compartmental approach. RESULTS: The results of this study suggested a significant genotypic as well as allelic association with valproic acid toxicity for UGT1A6 (541A>G) or UGT1A6 (552A>C) polymorphic enzymes. The elimination half-life (t 1/2 = 40.2 hr) of valproic acid was longer and the clearance rate (CL = 917 mL/hr) was lower in the poor metabolizers group of UGT1A6 (552A>C) polymorphism who showed toxicity than in the intermediate metabolizers group (t 1/2= 35.5 hr, CL = 1,022 mL/hr) or the extensive metabolizers group (t 1/2= 25.4 hr, CL = 1,404 mL/hr). CONCLUSION: /These/ findings suggest that the UGT1A6 (552A>C) genetic polymorphism plays a significant role in the steady state concentration of valproic acid, and it thereby has an impact on the toxicity of the valproic acid used in the patients with epilepsy. Biotransformation /of valproic acid/ is primarily hepatic. Some metabolites may have pharmacologic or toxic activity. Rate of metabolism is faster in children and in patients concurrently using enzyme-inducing medications, such as phenytoin, phenobarbital, primidone, and carbamazepine. Valproate is metabolized almost entirely by the liver. In adult patients on monotherapy, 30- 50% of an administered dose appears in urine as a glucuronide conjugate. Mitochondrial -oxidation is the other major metabolic pathway, typically accounting for over 40% of the dose. Usually, less than 15-20% of the dose is eliminated by other oxidative mechanisms. Less than 3% of an administered dose is excreted unchanged in urine. Valproic acid has known human metabolites that include 4-Hydroxyvalproate, 5-Hydroxyvalproate, (2S,3S,4S,5R)-3,4,5-Trihydroxy-6-(2-propylpentanoyloxy)oxane-2-carboxylic acid, 3-Hydroxyvalproate, and 4-ene-valproate. Valproic acid is rapidly absorbed from gastrointestinal tract. Valproic acid is metabolized almost entirely by the liver. In adult patients on monotherapy, 30-50% of an administered dose appears in urine as a glucuronide conjugate. Mitochondrial oxidation is the other major metabolic pathway, typically accounting for over 40% of the dose. These products include 2-n-propylpent-2-enoic acid (delta 2,3 VPE) and several coenzyme A (CoA) derivatives including VPA-CoA, and delta 2,3 VPE-CoA. Usually, less than 15-20% of the dose is eliminated by other oxidative mechanisms. Less than 3% of an administered dose is excreted unchanged in urine (A308). Half Life: 9-16 hours (following oral administration of 250 mg to 1000 mg). Biological Half-Life 13-19 hours. The half-life in neonates ranges from 10-67 hours while the half-life in pediatric patients under 2 months of age ranges from 7-13 hours. In children the half-life of valproic acid alone is 10 to 11 hours; when other medications are added, half-life may be reduced to 8 to 9 hours. Half-lives of up to 30 hours have been reported in overdosage. Variable, from 6 to 16 hours; may be considerably longer in patients with hepatic function impairment, in the elderly, and in children up to 18 months of age; may be considerably shorter in patients receiving hepatic enzyme-inducing anticonvulsants. In one study, the half life in children under 10 days ranged from 10 to 67 hours compared to a range of 7 to 13 hours in children greater than 2 months. |
| Toxicity/Toxicokinetics |
Toxicity Summary IDENTIFICATION AND USE: Valproic acid is a colorless to pale yellow viscous liquid. Valproic acid is an antiepileptic drug and is used solely or in combination with other anticonvulsants in the treatment of simple (petit mal) and complex absence seizures. Valproic acid may be effective against myoclonic and atonic seizures in young children. HUMAN EXPOSURE AND TOXICITY: After oral administration, the drug is rapidly absorbed from the gastrointestinal tract and metabolized in the liver. Fatal hepatic failure has been reported in patients on valproic acid therapy, especially those on chronic use. Pancreatitis has also been reported in patients receiving normal therapeutic dosage. Reports showed that acute toxicity is rare, and usually follows a benign course. The most commonly reported adverse effects are anorexia, nausea and vomiting. Central nervous system effects include drowsiness, possibly apathy and withdrawal, confusion, restlessness, hyperactivity. Less frequently, seizures and coma may occur. Sedative effects are more pronounced when drug is used together with other anti-epileptic agents. Hematopoietic system effects include thrombocytopenia, abnormal bleeding time and partial thromboplastin time with decreased fibrinogen levels and prolonged prothrombin time leading to bruising, petechiae, hematoma, and epistaxis. The drug can induce pruritic macular rashes and transient alopecia. Altered thyroid functions was described. Death is rare but if it occurs it results from cardiopulmonary arrest secondary to hepatic failure. Safe use of valproic acid during pregnancy has not been established. Although several reports suggest an association between the use of valproic acid in pregnant epileptic women and an increased incidence of birth defects (particularly neural tube defects) in children born to these women, a causal relationship remains to be established. The drug crosses the placental barrier and has been found in breast milk. The mechanism of action of valproic acid is unknown. Effects of the drug may be related, at least in part, to increased brain concentrations of the inhibitory neurotransmitter GABA. ANIMAL STUDIES: Animal studies have shown that valproic acid inhibits GABA transferase and succinic aldehyde dehydrogenase, enzymes which are important for GABA catabolism. Results of one study indicate the drug inhibits neuronal activity by increasing potassium conductance. In animals, valproic acid protects against seizure induced by electrical stimulation, as well as those induced by pentylenetetrazol. In 2 year rat and chronic mouse studies, an increased incidence of subcutaneous fibrosarcoma occurred in male rats at the higher dosage level and a dose related trend for an increased incidence of benign pulmonary adenomas was observed in male mice. The importance of these findings to humans is not known. Adverse fetal effects have been observed in reproduction studies in rats and mice. Studies have not shown any evidence of mutagenic potential for the drug. Valproic Acid binds to and inhibits GABA transaminase. This leads to increased brain concentrations of gamma-aminobutyric acid (GABA), an inhibitory neurotransmitter in the CNS. Acute poisoning by VPA can lead to severe CNS depression including coma, confusion, somnolence, dizziness or hallucinations. Hypotension, respiratory depression and hypo/hyperthermia are also common. VPA is also hepatotoxic, which is likely due to its mitochondrial toxicity. VPA appears to exert its mitochondrial toxicity by impairing mitochondrial functions leading to oxidative stress and cytochrome c expulsion, which leads to apoptosis (A15078). VPA is contraindicated in pregnancy due to its teratogenicity. VPA is a known folate antagonist, which can cause neural tube defects in developing fetuses. Thus, folic acid supplements in pregnant women may alleviate teratogenic problems associated with VPA use. VPA and its metabolites inhibit the biosynthesis of carnitine by decreasing the concentration of alpha-ketoglutarate (through direct inhibition of alpha-ketoglutarate dehydrogenase) and may contribute to carnitine deficiency. It is postulated that carnitine supplementation may increase the beta-oxidation of VPA, thereby limiting cytosolic omega-oxidation and the production of toxic metabolites that are involved in liver toxicity and ammonia accumulation. VPA-induced hepatotoxicity and hyperammonemic encephalopathy may be promoted either by a pre-existing carnitine deficiency or by deficiency induced by VPA per se. VPA has been shown to downregulate levels of superoxide dismutase (SOD), glutathione (GSH), histone deacetylase (HDAC) and folate. It has also been shown to upregulate H2O2 and homocysteine. Elevated levels of H2O2 negatively affect the NADPH reducing system for dihydrofolate reductase (DHFR) and methylene tetrahydrofolate reductase (MTHFR) (A15079). Toxicity Data Oral, mouse: LD50 = 1098 mg/kg; Oral, rat: LD50 = 670 mg/kg. In general, serum or plasma valproic acid concentrations are in a range of 20–100 mg/l during controlled therapy, but may reach 150–1500 mg/l following acute poisoning. Interactions Administration of a single oral 50 mg dose of amitriptyline to 15 normal volunteers (10 males and 5 females) who received valproate (500 mg BID) resulted in a 21% decrease in plasma clearance of amitriptyline and a 34% decrease in the net clearance of nortriptyline. Rare postmarketing reports of concurrent use of valproate and amitriptyline resulting in an increased amitriptyline level have been received. Concurrent use of valproate and amitriptyline has rarely been associated with toxicity. Monitoring of amitriptyline levels should be considered for patients taking valproate concomitantly with amitriptyline. Consideration should be given to lowering the dose of amitriptyline/nortriptyline in the presence of valproate. A clinically significant reduction in serum valproic acid concentration has been reported in patients receiving carbapenem antibiotics (for example, ertapenem, imipenem, meropenem; this is not a complete list) and may result in loss of seizure control. The mechanism of this interaction is not well understood. Serum valproic acid concentrations should be monitored frequently after initiating carbapenem therapy. Alternative antibacterial or anticonvulsant therapy should be considered if serum valproic acid concentrations drop significantly or seizure control deteriorates... Serum levels of carbamazepine (CBZ) decreased 17% while that of carbamazepine-10,11-epoxide (CBZ-E) increased by 45% upon co-administration of valproate and CBZ to epileptic patients. A study involving the co-administration of 1200 mg/day of felbamate with valproate to patients with epilepsy (n = 10) revealed an increase in mean valproate peak concentration by 35% (from 86 to 115 mcg/mL) compared to valproate alone. Increasing the felbamate dose to 2400 mg/day increased the mean valproate peak concentration to 133 mcg/mL (another 16% increase). A decrease in valproate dosage may be necessary when felbamate therapy is initiated. For more Interactions (Complete) data for VALPROIC ACID (61 total), please visit the HSDB record page. Non-Human Toxicity Values LD50 Guinea pig oral 824 mg/kg LD50 Mouse sc 860 mg/kg LD50 Mouse ip 470 mg/kg LD50 Mouse oral 1098 mg/kg For more Non-Human Toxicity Values (Complete) data for VALPROIC ACID (7 total), please visit the HSDB record page. |
| References |
[1]. Valproic acid inhibits the growth of HeLa cervical cancer cells via caspase-dependent apoptosis. Oncol Rep. 2013 Dec;30(6):2999-3005. [2]. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001 Sep 28;276(39):36734-41. [3]. Valproic acid inhibits tumor angiogenesis in mice transplanted with Kasumi 1 leukemia cells. Mol Med Rep. 2014 Feb;9(2):443-9. [4]. Acute prenatal exposure to a moderate dose of valproic acid increases social behavior and alters gene expression in rats. Int J Dev Neurosci. 2013 Dec;31(8):740-50. [5]. Valproic Acid Is a Novel Activator of AMP-Activated Protein Kinase and Decreases Liver Mass, Hepatic Fat Accumulation, and Serum Glucose in Obese Mice. Mol Pharmacol. 2014 Jan;85(1):1-10. [6]. Valproic acid induces Notch1 signaling in small cell lung cancer cells. J Surg Res. 2008 Jul;148(1):31-7. [7]. Valproic acid in association with highly active antiretroviral therapy for reducing systemic HIV-1 reservoirs: results from a multicentre randomized clinical study. HIV Med. 2012 May;13(5):291-6. |
| Additional Infomation |
Therapeutic Uses /EXP THER/ Emerging evidence suggests that adenomyosis, like endometriosis, may also be an epigenetic disease. In this study, we evaluated the effect of valproic acid (VPA) in ICR mice with adenomyosis, induced by neonatal dosing with tamoxifen. For all mice, we evaluated the bodyweight and the response to thermal stimuli by hotplate and tail-flick tests 4, 8, and 12 weeks after dosing, respectively, and then treated mice with low- and high-dose of VPA, progesterone (P4), P4 + VPA, or vehicle only. Three weeks after treatment, both bodyweight and thermal response tests were evaluated again before sacrifice, and the depth of myometrial infiltration was evaluated. We found that: (i) the induction of adenomyosis resulted in progressive generalized hyperalgesia as measured by hotplate and tail-flick tests, along with decreased bodyweight; (ii) treatment with VPA, P4, or a combination was efficacious in improving generalized hyperalgesia; and (iii) drug treatment appeared to reduce the myometrial infiltration, but the difference did not reach statistical significance. Thus, VPA seems to be a promising therapeutics for treating adenomyosis, as reported recently in some case series in humans. /EXP THER/ Purpose: 5-Azacytidine (5-AZA) is a DNA-hypomethylating agent. Valproic acid is a histone deacetylase inhibitor. Combining hypomethylating agents and histone deacetylase inhibitors produces synergistic anticancer activity in vitro and in vivo. On the basis of this evidence, /this study/ conducted a phase I study of the combination of 5-AZA and valproic acid in patients with advanced cancers. Experimental Design: 5-AZA was administered s.c. daily for 10 days. Valproic acid was given orally daily with a goal to titrate to plasma levels of 75 to 100 mug/mL (therapeutic for seizures). Cycles were 28 days long. 5-AZA was started at 20 mg/m(2) and escalated using an adaptive algorithm based on the toxicity profile in the prior cohort (6 + 6 design). Peripheral blood mononuclear cell global DNA methylation and histone H3 acetylation were estimated with the long interspersed nucleotide elements pyrosequencing assay and Western blots, respectively, on days 1 and 10 of each cycle when patients agreed to provide them. Results: Fifty-five patients were enrolled. Median age was 60 years (range, 12-77 years). The maximum tolerated dose was 75 mg/m(2) of 5-AZA in combination with valproic acid. Dose-limiting toxicities were neutropenic fever and thrombocytopenia, which occurred at a dose of 94 mg/m(2) of 5-AZA. Stable disease lasting 4 to 12 months (median, 6 months) was observed in 14 patients (25%). A significant decrease in global DNA methylation and induction of histone acetylation were observed. Conclusion: The combination of 5-AZA and valproic acid is safe at doses up to 75 mg/m(2) for 5-AZA in patients with advanced malignancies. /EXP THER/ Amphetamine (AMPH)-induced hyper-locomotion has been well manifested in an animal model of psychiatric diseases such as drug addiction and bipolar disorder. /This study/ investigated the effects on AMPH-induced locomotor activity of chronically microinjected valproic acid into the nucleus accumbens (NAcc). Rats with guide cannular implanted bilaterally were divided into three groups and either saline or valproic acid (100 or 300 ug/0.5 uL/side) was microinjected into the NAcc once daily for 7 days. On day 8, half of each group received either saline or AMPH (1mg/kg, i.p.), respectively, and locomotor activity was measured for 2 hr. The increases of both horizontal locomotion and rearing by AMPH were attenuated in the rat pre-treated with valproic acid compared to saline in a dose-dependent manner. These results indicate that neuronal modifications in the NAcc induced by chronic valproic acid can modulate amphetamine-induced locomotor activity. /EXP THER/ Purpose: Non-small-cell lung cancer (NSCLC) accounts for the majority of lung cancer and is the most common cause of cancer death in industrialized countries. Epigenetic modifications are observed universally during the tumorigenesis of lung cancer. The development of epigenetic-modulating agents utilizing the synergism between hypomethylating agents and histone deacetylase (HDAC) inhibitors provides a novel therapeutic approach in treating NSCLC. Methods: /This study/ performed a phase I trial combining 5-aza-2'-deoxycytidine (decitabine) and valproic acid (VPA), in patients with advanced stage NSCLC. Patients were treated with escalating doses of decitabine (5-15 mg/sq m) IV for 10 days in combination with VPA (10-20 mg/kg/day) PO on days 5-21 of a 28-day cycle. Pharmacokinetic and pharmacodynamic analysis included decitabine pharmacokinetics and fetal hemoglobin expression. Results: Eight patients were accrued to this phase I study. All patients had advanced NSCLC and had received prior chemotherapy. Eastern Cooperative Oncology Group performance status was 0-2. Major toxicities included myelosuppression and neurotoxicity. Dose-limiting toxicity was seen in two patients suffering grade 3 neurotoxicity during cycle one including disorientation, lethargy, memory loss, and ataxia at dose level 1. One patient had grade 3 neutropenia at the de-escalated dose. No objective response was observed, and stable disease was seen in one patient. Fetal hemoglobin levels increased after cycle one in all seven patients with evaluable results. Conclusions: /This study/ observed that decitabine and valproic acid are an effective combination in reactivating hypermethylated genes as demonstrated by re-expressing fetal hemoglobin. This combination in patients with advanced stage IV NSCLC, however, is limited by unacceptable neurological toxicity at a relatively low dosage. Combining hypomethylating agents with alternative HDAC inhibitors that lack the toxicity of VPA should be explored further. For more Therapeutic Uses (Complete) data for VALPROIC ACID (8 total), please visit the HSDB record page. Drug Warnings /BOXED WARNING/ WARNING: LIFE THREATENING ADVERSE REACTIONS. Hepatotoxicity: General Population: Hepatic failure resulting in fatalities has occurred in patients receiving valproate. These incidents usually have occurred during the first six months of treatment. Serious or fatal hepatotoxicity may be preceded by non-specific symptoms such as malaise, weakness, lethargy, facial edema, anorexia, and vomiting. In patients with epilepsy, a loss of seizure control may also occur. Patients should be monitored closely for appearance of these symptoms. Serum liver tests should be performed prior to therapy and at frequent intervals thereafter, especially during the first six months. Children under the age of two years are at a considerably increased risk of developing fatal hepatotoxicity, especially those on multiple anticonvulsants, those with congenital metabolic disorders, those with severe seizure disorders accompanied by mental retardation, and those with organic brain disease. When Depakene products are used in this patient group, they should be used with extreme caution and as a sole agent. The benefits of therapy should be weighed against the risks. The incidence of fatal hepatotoxicity decreases considerably in progressively older patient groups. Patients with Mitochondrial Disease: There is an increased risk of valproate-induced acute liver failure and resultant deaths in patients with hereditary neurometabolic syndromes caused by DNA mutations of the mitochondrial DNA Polymerase gamma (POLG) gene (e.g. Alpers Huttenlocher Syndrome). Depakene is contraindicated in patients known to have mitochondrial disorders caused by POLG mutations and children under two years of age who are clinically suspected of having a mitochondrial disorder. In patients over two years of age who are clinically suspected of having a hereditary mitochondrial disease, Depakene should only be used after other anticonvulsants have failed. This older group of patients should be closely monitored during treatment with Depakene for the development of acute liver injury with regular clinical assessments and serum liver testing. POLG mutation screening should be performed in accordance with current clinical practice. Fetal Risk: Valproate can cause major congenital malformations, particularly neural tube defects (e.g., spina bifida). In addition, valproate can cause decreased IQ scores following in utero exposure. Valproate should only be used to treat pregnant women with epilepsy if other medications have failed to control their symptoms or are otherwise unacceptable. Valproate should not be administered to a woman of childbearing potential unless the drug is essential to the management of her medical condition. This is especially important when valproate use is considered for a condition not usually associated with permanent injury or death (e.g., migraine). Women should use effective contraception while using valproate. Pancreatitis: Cases of life-threatening pancreatitis have been reported in both children and adults receiving valproate. Some of the cases have been described as hemorrhagic with a rapid progression from initial symptoms to death. Cases have been reported shortly after initial use as well as after several years of use. Patients and guardians should be warned that abdominal pain, nausea, vomiting, and/or anorexia can be symptoms of pancreatitis that require prompt medical evaluation. If pancreatitis is diagnosed, valproate should ordinarily be discontinued. Alternative treatment for the underlying medical condition should be initiated as clinically indicated. Because of these changes in valproate clearance, monitoring of valproate and concomitant drug concentrations should be increased whenever enzyme inducing drugs are introduced or withdrawn. Since valproate may interact with concurrently administered drugs which are capable of enzyme induction, periodic plasma concentration determinations of valproate and concomitant drugs are recommended during the early course of therapy. Valproate is partially eliminated in the urine as a keto-metabolite which may lead to a false interpretation of the urine ketone test. For more Drug Warnings (Complete) data for VALPROIC ACID (58 total), please visit the HSDB record page. Pharmacodynamics Valproate has been shown to reduce the incidence of complex partial seizures and migraine headaches. It also improves symptom control in bipolar mania. Although the exact mechanisms responsible are unknown, it is thought that valproate produces increased cortical inhibition to contribute to control of neural synchrony. It is also thought that valproate exerts a neuroprotective effect preventing damage and neural degeneration in epilepsy, migraines, and bipolar disorder. Valproate is hepatotoxic and teratogenic. The reasons for this are unclear but have been attributed to the genomic effects of the drug. A small proof-of concept study found that valproate increases clearance of human immunodeficiency virus (HIV) when combined with highly active antiretroviral therapy (HAART) by reactivating the virus to allow clearance, however, a larger multicentre trial failed to show a significant effect on HIV reservoirs when added to HAART. The FDA labeling contains a warning regarding HIV reactivation during valproate use.. |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (17.34 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (17.34 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (17.34 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 2 mg/mL (13.87 mM) in PBS (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. Solubility in Formulation 5: 20 mg/mL (138.69 mM) in 0.5% CMC/saline water (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 6.9343 mL | 34.6717 mL | 69.3433 mL | |
| 5 mM | 1.3869 mL | 6.9343 mL | 13.8687 mL | |
| 10 mM | 0.6934 mL | 3.4672 mL | 6.9343 mL |