Physicochemical Properties
| Molecular Formula | C26H26N2O5 |
| Molecular Weight | 446.503 |
| Exact Mass | 446.184 |
| Elemental Analysis | C, 69.94; H, 5.87; N, 6.27; O, 17.92 |
| CAS # | 955371-66-1 |
| PubChem CID | 24802973 |
| Appearance | White to off-white solid powder |
| Density | 1.3±0.1 g/cm3 |
| Boiling Point | 698.6±55.0 °C at 760 mmHg |
| Flash Point | 376.3±31.5 °C |
| Vapour Pressure | 0.0±2.2 mmHg at 25°C |
| Index of Refraction | 1.644 |
| LogP | 4.25 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 8 |
| Heavy Atom Count | 33 |
| Complexity | 648 |
| Defined Atom Stereocenter Count | 0 |
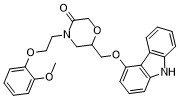
| SMILES | O=C1COC(COC2C3=C(NC4C3=CC=CC=4)C=CC=2)CN1CCOC1C(OC)=CC=CC=1 |
| InChi Key | OPUVSUMPCOUABG-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C26H26N2O5/c1-30-22-10-4-5-11-23(22)31-14-13-28-15-18(32-17-25(28)29)16-33-24-12-6-9-21-26(24)19-7-2-3-8-20(19)27-21/h2-12,18,27H,13-17H2,1H3 |
| Chemical Name | 6-[(9H-Carbazol-4-yloxy)methyl]-4-[2-(2-methoxyphenoxy)ethyl]-3-morpholinone |
| Synonyms | VK-II 36VK-II-36 VK-II36 VKII36 VK II36 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
L-type calcium channel (ICa,L) (IC50 = 3.2 μM in rabbit ventricular myocytes) [1] Rapidly activating delayed rectifier potassium channel (IKr/hERG) (IC50 = 7.8 μM in HEK293 cells expressing hERG) [1] β1-adrenergic receptor [1] |
| ln Vitro |
Inhibition of EAD-induced triggered activities: In isolated rabbit ventricular myocytes (EAD model: low K⁺/high Ca²⁺ perfusion), VK-II-36 (1–10 μM) dose-dependently inhibits EADs and associated triggered beats. At 5 μM, EAD incidence is reduced from 82% (control) to 21%, and triggered activity frequency decreases by 75% (patch-clamp recording) [1] - Inhibition of DAD-induced triggered activities: In rabbit ventricular myocytes (DAD model: ouabain-induced Na⁺/K⁺ ATPase inhibition), VK-II-36 (2–15 μM) suppresses DADs and diastolic calcium oscillations. At 10 μM, DAD amplitude is reduced by 68%, and triggered activity is abolished in 70% of cells [1] - Modulation of ion currents: The compound inhibits L-type calcium current (ICa,L) with an IC50 of 3.2 μM, and hERG-mediated IKr with an IC50 of 7.8 μM (whole-cell patch-clamp). It shows weak inhibition of Na⁺ current (INa) (< 20% inhibition at 10 μM) [1] - Action potential (AP) regulation: In human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs), VK-II-36 (5 μM) shortens APD90 (action potential duration at 90% repolarization) by 22% without prolonging QT interval (a marker of arrhythmia risk) [1] - Calcium overload attenuation: In ouabain-treated myocytes, VK-II-36 (10 μM) reduces cytosolic calcium overload by 55% (fura-2 AM calcium imaging), preventing DAD formation [1] |
| ln Vivo |
Background: Carvedilol and its analogues suppress delayed afterdepolarizations (DADs) and catecholaminergic polymorphic ventricular tachycardias by direct action on the cardiac ryanodine receptor type 2 (RyR2). Objective: To test a hypothesis that carvedilol analogue may also prevent triggered activities (TAs) through the suppression of early afterdepolarizations (EADs). Methods: Intracellular Ca(2+) and membrane voltage were simultaneously recorded by using optical mapping technique in Langendorff-perfused mouse and rabbit hearts to study the effect of carvedilol analogue VK-II-36, which does not have significant beta-blocking effects. Results: Spontaneous intracellular Ca(2+) elevations (SCaEs) during diastole were induced by rapid ventricular pacing and isoproterenol infusion in intact rabbit ventricles. Systolic and diastolic SCaEs were simultaneously noted in Langendorff-perfused RyR2 R4496(+/-) mouse hearts after creating atrioventricular block. VK-II-36 effectively suppressed SCaEs and eliminated TAs observed in both mouse and rabbit ventricles. We tested the effect of VK-II-36 on EADs by using a rabbit model of acquired long QT syndrome, in which phase 2 and phase 3 EADs were observed in association with systolic SCaEs. VK-II-36 abolished the systolic SCaEs and phase 2 EADs, and greatly decreased the dispersion of repolarization and the amplitude of phase 3 EADs. VK-II-36 completely prevented EAD-mediated TAs in all ventricles studied. Conclusions: A carvedilol analogue, VK-II-36, inhibits ventricular tachyarrhythmias in intact mouse and rabbit ventricles by the suppression of SCaEs, independent of beta-blocking activity. The RyR2 may be a potential target for treating focal ventricular arrhythmias triggered by either EADs or DADs.[1] VK-II-36 suppresses DAD-mediated arrhythmias That diastolic SCaEs can be induced by a prolonged rapid ventricular pacing (cycle length 200 ms for 200 beats) under isoproterenol infusion (0.01 to 0.3 μM) in the rabbit ventricle. Since diastolic SCaEs represent the sum of the Ca2+ waves at the tissue level, this rabbit model was used to determine whether VK-II-36 prevents DAD-mediated ventricular arrhythmias (n = 12). Optical mapping of Cai revealed that diastolic SCaEs were observed following prolonged rapid ventricular pacing under isoproterenol infusion in all 12 hearts studied . VK-II-36 (30 μM) suppressed the diastolic SCaE (0.139 ± 0.017 AU to 0.007 ± 0.004 AU, P < 0.001), and DAD (0.035 ± 0.011 AU to 0.00 ± 0.00 AU, P = 0.008). Ventricular arrhythmias were reproducibly induced in 7 of 12 hearts (single TAs in 2 hearts, VTs in 5 hearts). VK-II-36 (30 μM) abolished all episodes of the ventricular arrhythmias . Thus, VK-II-36 effectively prevented DAD-mediated ventricular arrhythmias in intact rabbit hearts.[1] VK-II-36 suppresses EAD-mediated arrhythmias and reduces dispersion of repolarization VK-II-36 (30 μM) consistently eliminated the systolic SCaEs (0.08 ± 0.03 AU to 0.00 ± 0.00 AU, P = 0.04) and phase-2 EADs (0.03 ± 0.01 AU to 0.00 ± 0.00 AU, P = 0.067). Also, VK-II-36 decreased phase-3 EADs (0.23 ± 0.02 AU to 0.05 ± 0.02 AU, P = 0.003). Since phase-2 EADs developed only at sites with a long APD, the elimination of phase-2 EADs shortened the maximal APD70 (458 ± 37 ms to 310 ± 19 AU, P = 0.002), but did not significantly change the minimal APD70 (263 ± 12 ms to 244 ± 10 ms, P = 0.10). As a result, VK-II-36 dramatically reduced the spatial dispersion of repolarization (APD70 dispersion: 195 ± 33 ms to 66 ± 14 ms, P = 0.002; standard deviation of APD70: 48 ± 10 ms to 15 ± 4 ms, P = 0.005). The decrease in the spatial dispersion of repolarization was accompanied by a decrease in Vm gradient during repolarization (0.33 ± 0.02 AU/mm to 0.18 ± 0.03 AU/mm, P = 0.003), which accounts for the suppression of phase-3 EADs since electrotonic depolarization across a high Vm gradient underlies the mechanism of phase-3 EAD.15 With Ikr blockade and 50% reduction in extracellular K+ and Mg2+, both phase-2 and phase-3 EAD developed TAs leading to polymorphic VT . VK-II-36 (30 μM) abolished the TAs and VTs induced by EADs in all hearts studied . Therefore, VK-II-36 directly abolished phase-2 EADs with indirect suppression of phase-3 EADs, which eliminated the EAD-mediated ventricular arrhythmias in a rabbit model of acquired long QT syndrome.[1] VK-II-36 suppresses both systolic and diastolic SCaEs VK-II-36 (10 μM) suppressed both systolic and diastolic SCaEs (0.14 ± 0.03 AU to 0.00 ± 0.00 AU, and 0.23 ± 0.02 AU to 0.11 ± 0.01 AU, respectively, both P < 0.05), indicating that carvedilol analogue has the potential for the treatment of cardiac arrhythmias associated with TA induced by either EADs or DADs. EAD-related arrhythmia inhibition (low K⁺ model): Rats were subjected to low-K⁺ (2.5 mM) perfusion to induce EAD-mediated ventricular arrhythmias. Intravenous administration of VK-II-36 (1 mg/kg, 3 mg/kg) reduced ventricular premature beats (VPBs) by 45% and 72%, respectively, and prevented ventricular tachycardia (VT) in 60% of rats (3 mg/kg dose) [1] - DAD-related arrhythmia inhibition (ouabain model): Dogs were treated with ouabain (40 μg/kg, i.v.) to induce DAD-mediated arrhythmias. VK-II-36 (2 mg/kg, i.v.) administered 15 minutes post-ouabain reduced VT incidence from 90% (control) to 30%, and improved survival rate from 40% to 80% within 2 hours [1] - Myocardial ischemia-reperfusion arrhythmia suppression: Rats underwent 30-minute myocardial ischemia followed by 2-hour reperfusion. VK-II-36 (3 mg/kg, i.v.) given 5 minutes before reperfusion reduced reperfusion-induced VPBs by 65% and VT/VF (ventricular fibrillation) incidence by 58% [1] - Hemodynamic effects: In anesthetized dogs, VK-II-36 (2 mg/kg, i.v.) caused a mild decrease in systolic blood pressure (12%) and heart rate (8%), with no significant hypotension or bradycardia (vs. carvedilol’s 20% BP reduction) [1] |
| Enzyme Assay |
hERG channel current assay: HEK293 cells stably expressing hERG were cultured and patched using whole-cell patch-clamp technique. The extracellular solution contained standard electrolytes, and intracellular solution included ATP. Serial dilutions of VK-II-36 (0.1 μM–50 μM) were applied, and IKr was recorded during voltage steps (from -80 mV to +40 mV, holding potential -70 mV). Current amplitude was quantified, and IC50 was derived from dose-response curves [1] - L-type calcium current (ICa,L) assay: Isolated rabbit ventricular myocytes were patched in whole-cell mode. ICa,L was elicited by depolarizing steps (from -40 mV to +60 mV, holding potential -80 mV) before and after VK-II-36 (0.5 μM–30 μM) application. Peak ICa,L amplitude was measured, and inhibition rate was calculated relative to baseline [1] |
| Cell Assay |
Ventricular myocyte isolation and EAD/DAD induction: Rabbit ventricular myocytes were isolated via collagenase digestion. For EAD induction, cells were perfused with low-K⁺ (2.5 mM) and high-Ca²⁺ (3.6 mM) Tyrode’s solution. For DAD induction, ouabain (1 μM) was added to standard Tyrode’s solution. VK-II-36 (1–15 μM) was perfused for 10 minutes, and triggered activities were recorded via patch-clamp (current-clamp mode) [1] - Action potential recording in hiPSC-CMs: hiPSC-CMs were cultured on glass coverslips and patched in current-clamp mode. Action potentials were elicited by 2-ms current pulses (1.5× threshold). VK-II-36 (1–10 μM) was applied, and APD50, APD90, and resting membrane potential were measured over 15 minutes [1] - Calcium imaging assay: Isolated myocytes were loaded with fura-2 AM (5 μM) for 30 minutes at 37°C. Cytosolic calcium concentration ([Ca²⁺]i) was measured via fluorescence microscopy (excitation 340/380 nm, emission 510 nm). VK-II-36 (5–15 μM) was added to ouabain-treated cells, and [Ca²⁺]i oscillations were quantified [1] |
| Animal Protocol |
Low-K⁺-induced EAD arrhythmia model (rats): Male Sprague-Dawley rats (250–300 g, n=8 per group) were anesthetized, and a jugular vein catheter was inserted for drug administration. The heart was exposed via thoracotomy, and a suction electrode was placed on the ventricular surface to record electrograms. Rats were perfused with low-K⁺ Tyrode’s solution (2.5 mM KCl) to induce arrhythmias. VK-II-36 (1 mg/kg, 3 mg/kg) or vehicle was injected intravenously, and arrhythmias were recorded for 60 minutes [1] - Ouabain-induced DAD arrhythmia model (dogs): Beagle dogs (10–15 kg, n=6 per group) were anesthetized, intubated, and instrumented with ECG leads and arterial catheters. Ouabain (40 μg/kg, i.v.) was administered to induce ventricular arrhythmias. Fifteen minutes later, VK-II-36 (2 mg/kg, i.v.) or vehicle was injected, and ECG and hemodynamics (blood pressure, heart rate) were monitored for 2 hours [1] - Myocardial ischemia-reperfusion model (rats): Male Wistar rats (200–250 g, n=7 per group) were anesthetized, and the left anterior descending coronary artery was ligated for 30 minutes (ischemia) followed by suture release (reperfusion). VK-II-36 (3 mg/kg, i.v.) or vehicle was injected 5 minutes before reperfusion. ECG was recorded continuously for 2 hours, and arrhythmias were classified per Lambeth Conventions [1] |
| Toxicity/Toxicokinetics |
Acute hemodynamic safety: In anesthetized rats and dogs, VK-II-36 doses up to 5 mg/kg (i.v.) did not cause severe hypotension (systolic BP < 80 mmHg) or bradycardia (heart rate < 250 bpm in rats, < 60 bpm in dogs) [1] - QT interval effect: In dogs, VK-II-36 (2 mg/kg, i.v.) did not prolong QT interval (ΔQT < 10 ms) compared to vehicle, indicating low risk of torsades de pointes [1] - Plasma protein binding: In vitro assay showed VK-II-36 binds to human plasma proteins at a rate of 88% [1] |
| References |
[1]. Carvedilol analogue inhibits triggered activities evoked by both early and delayed afterdepolarizations. Heart Rhythm. 2013 Jan;10(1):101-7. |
| Additional Infomation |
Background: Triggered activities evoked by EADs and DADs are key mechanisms underlying life-threatening ventricular arrhythmias (e.g., during myocardial ischemia, heart failure, drug toxicity). Current antiarrhythmics often have limited efficacy or proarrhythmic risks, making novel agents targeting both EADs and DADs valuable [1] - Mechanism of action: VK-II-36 exerts dual effects: 1) Inhibits ICa,L to reduce calcium overload (preventing EADs and DADs); 2) Moderately blocks IKr to normalize repolarization without excessive QT prolongation. Unlike carvedilol, it has weaker β-adrenergic blocking activity, minimizing hemodynamic side effects [1] - Therapeutic potential: The compound shows promise for treating arrhythmias associated with EADs/DADs (e.g., ischemia-reperfusion arrhythmias, digitalis toxicity, long QT syndrome). Its favorable safety profile (mild hemodynamic effects, no QT prolongation) supports further development as an antiarrhythmic agent [1] - Chemical feature: As a carvedilol analogue, VK-II-36 retains the phenylethanolamine scaffold but with modified substitutions, enhancing selectivity for ion channels over β-receptors. It has good solubility in DMSO (≥ 10 mM) and moderate aqueous solubility (1.1 mg/mL in pH 7.4 buffer) [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~100 mg/mL (~223.96 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.60 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.60 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2396 mL | 11.1982 mL | 22.3964 mL | |
| 5 mM | 0.4479 mL | 2.2396 mL | 4.4793 mL | |
| 10 mM | 0.2240 mL | 1.1198 mL | 2.2396 mL |