Ulecaciclib is a novel, potent and orally bioactive inhibitor of cyclin-dependent kinase (CDK) with antitumor effects. It inhibits CDK with Ki values of 0.62 μM (CDK2/Cyclin A), 0.2 nM (CDK4/Cyclin D1), 3 nM (CDK6/Cyclin D3), and 0.63 μM (CDK7/Cyclin H), respectively.
Physicochemical Properties
| Molecular Formula | C25H33FN8S |
| Molecular Weight | 496.646526098251 |
| Exact Mass | 496.253 |
| CAS # | 2075750-05-7 |
| PubChem CID | 126535127 |
| Appearance | Typically exists as solid at room temperature |
| LogP | 4.2 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 10 |
| Rotatable Bond Count | 8 |
| Heavy Atom Count | 35 |
| Complexity | 647 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | POFVJRKJJBFPII-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C25H33FN8S/c1-3-33-10-12-34(13-11-33)16-18-8-9-21(27-14-18)31-24-28-15-20(26)22(32-24)23-17(2)29-25(35-23)30-19-6-4-5-7-19/h8-9,14-15,19H,3-7,10-13,16H2,1-2H3,(H,29,30)(H,27,28,31,32) |
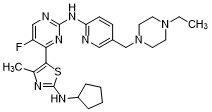
| Chemical Name | N-cyclopentyl-5-[2-[[5-[(4-ethylpiperazin-1-yl)methyl]pyridin-2-yl]amino]-5-fluoropyrimidin-4-yl]-4-methyl-1,3-thiazol-2-amine |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| ln Vitro | Compound 2, ulecaciclib, exhibits an inhibitory impact on CDK kinase, as demonstrated by its Ki values of 0.62 μM (CDK2/A), 0.2 nM (CDK4/Cyclin D1), 3 nM (CDK6/Cyclin D3), and 0.63 μM (CDK7/H), respectively, after 40 minutes of exposure[2]. Ulecaciclib (72 h) has a growth inhibition GI50 value of 10 nM and has a high antiproliferative action against leukemia cells[2]. Ulecaciclib (72 hours) specifically inhibits Ovarian A2780 with a GI50 value of 40 nM and suppresses tumor growth with GI50s ranging from 0.04-5.09 μM[2]. |
| ln Vivo | In mice, ulecaciclib (compound 2), at 2 mg/kg for intravenous administration and 10 mg/kg for po administration, exhibits a noteworthy inclination to cross the blood-brain barrier, with brain/plasma ratios of >1.2 (iv) and >0.7 (po), respectively[2]. Mice treated with ulecaciclib (200 mg/kg po daily for 21 days) exhibit in vivo anti-tumor efficacy[2]. When combined with TMZ (5 mg/kg; po; 5 d/week; 2 weeks), ulecaciclib (25 mg/kg; po; daily; 10 d) exhibits strong anti-tumor efficacy at lower doses in mice[2]. In male cynomolgus monkeys, ulecaciclib (compound A) (50 mg/kg; po) exhibits a good pharmacokinetic profile with a half-life of 8.34 h and a Tmax of 6.67 h, as well as Cmax = 643 ng/mL and AUC(0-24) = 9543 ng·h/mL. Cynomolgus monkey pharmacokinetics of ulecaciclib[3] Route Dose (mg/kg) T1/2 (h) Tmax (h) Cmax (ng/mL) AUC(0-t) (h·ng/mL) AUC(0-∞) (h·ng/mL) Vd (L/kg) CL (mL/min/kg) MRT(0-t) (h) F (%) iv 5 6.53 / 447 4187 4560 9.79 19.4 6.64 / po 50 8.34 6.67 643 9543 7305 / / 10.4 21.8 |
| Cell Assay |
Cell Viability Assay[2] Cell Types: U87, U251, T98G (mycoplasma-free); and MB453, Colo205, H460, A2780, PANC1, LNC, M229 Tested Concentrations: 0-10 μM Incubation Duration: 72 hrs (hours) Experimental Results: Inhibited cancer cells growth with GI50s of 2.17 μM (U87), 5.09 μM (U251), 4.18 μM (T98G), 0.62 μM (MB453), 1.55 μM (Colo205 ), 0.41 μM (H460), 0.04 μM (A2780), 1.21 μM (PANC1), 0.28 μM (LNC), 0.83 μM (M229). |
| Animal Protocol |
Animal/Disease Models: CDl nu/nu female mice (5-6 weeks old; injected with U87 GBM cells, sc)[2] Doses: 200 mg/kg Route of Administration: po (oral gavage); daily; 21 days Experimental Results: decreased tumor growth markedly without any overt toxicity. Animal/Disease Models: GBM orthotopic mouse xenograft models[2] Doses: 120 mg/kg Route of Administration: po (oral gavage); daily for 2 days Experimental Results: Inhibited tumor growth on day 21 and increased life span ratio (ILS) of 154.8% for teated mice. ILS = (DaysT - DaysC)/DaysC, where DaysC = days survived by control group and DaysT = days survived by treatment group. |
| References |
[1]. International Nonproprietary Names for Pharmaceutical Substances (INN). WHO Drug Information. 2022. 36(2):337. [2]. Treatment of proliferative diseases of the CNS using a class of thiazole-pyrimidine compounds that inhibit the activity of CDK4 and/or CDK6[P]. World Intellectual Property Organization, WO2021222967 A1 2021-11-11. [3]. Succinate and crystal form thereof as therapeutics[P]. World Intellectual Property Organization, WO2022099357 A1 2022-05-19. |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0135 mL | 10.0675 mL | 20.1349 mL | |
| 5 mM | 0.4027 mL | 2.0135 mL | 4.0270 mL | |
| 10 mM | 0.2013 mL | 1.0067 mL | 2.0135 mL |