Tipranavir (PNU140690; PNU-140690; trade name: Aptivus) is a marketed anti-HIV drug acting as a nonpeptidic multi-protease inhibitor (IC50s = 66-410 nM). Tipranavir is able to inhibit the replication of viruses that are resistant to other protease inhibitors and it recommended for patients who are resistant to other treatments. Resistance to tipranavir itself seems to require multiple mutations. Tipranavir was approved by the Food and Drug Administration (FDA) on June 22, 2005, and was approved for pediatric use on June 24, 2008. Like lopinavir and atazanavir, it is very potent and is effective in salvage therapy for patients with some drug resistance.
Physicochemical Properties
| Molecular Formula | C31H33N2O5F3S |
| Molecular Weight | 602.66432 |
| Exact Mass | 602.206 |
| CAS # | 174484-41-4 |
| Related CAS # | Tipranavir-d4;1217819-15-2 |
| PubChem CID | 54682461 |
| Appearance | White to off-white solid powder |
| Density | 1.313g/cm3 |
| Boiling Point | 680ºC at 760mmHg |
| Melting Point | 86-89ºC |
| Flash Point | 365.1ºC |
| Vapour Pressure | 0mmHg at 25°C |
| Index of Refraction | 1.579 |
| LogP | 8.479 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 10 |
| Rotatable Bond Count | 11 |
| Heavy Atom Count | 42 |
| Complexity | 1050 |
| Defined Atom Stereocenter Count | 2 |
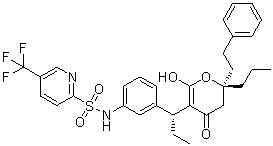
| SMILES | CCC[C@]1(CC(=C(C(=O)O1)[C@H](CC)C2=CC(=CC=C2)NS(=O)(=O)C3=NC=C(C=C3)C(F)(F)F)O)CCC4=CC=CC=C4 |
| InChi Key | SUJUHGSWHZTSEU-FYBSXPHGSA-N |
| InChi Code | InChI=1S/C31H33F3N2O5S/c1-3-16-30(17-15-21-9-6-5-7-10-21)19-26(37)28(29(38)41-30)25(4-2)22-11-8-12-24(18-22)36-42(39,40)27-14-13-23(20-35-27)31(32,33)34/h5-14,18,20,25,36-37H,3-4,15-17,19H2,1-2H3/t25-,30-/m1/s1 |
| Chemical Name | N-[3-[(1R)-1-[(2R)-4-hydroxy-6-oxo-2-(2-phenylethyl)-2-propyl-3H-pyran-5-yl]propyl]phenyl]-5-(trifluoromethyl)pyridine-2-sulfonamide |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
IC50: 66-410 nM (HIV-1 isolates)[1] HIV-1 protease (PR) dimerization and enzymatic activity inhibition. [1] |
| ln Vitro |
Tipranavir (PNU-140690) exhibits strong action against a broad range of wild-type and multi-PI-resistant HIV-1 variants, inhibits the enzymatic activity of HIV-1 protease, and prevents the dimerization of protease subunits. HIV11MIX, a mixture of 11 multi-PI-resistant (but TPV-sensitive) clinical isolates (including HIVB and HIVC), quickly develops high-level Tipranavir (PNU-140690) resistance and replicates at high concentrations of Tipranavir (PNU-140690) after being selected against Tipranavir (by 10 passages [HIV11MIXP10]). With IC50s of 2.9 and 3.2 μM, respectively, which represent 11- and 12-fold increases in comparison to the IC50 against cHIVB, cHIVBI54V and cHIVBI54V/V82T are considerably resistant to Tipranavir (PNU-140690)[1]. Tipranavir exerts potent activity against multi-PI-resistant HIV-1 isolates. A mixture of 11 multi-PI-resistant clinical isolates (HIV11MIX) was selected against TPV, and by passage 10 (HIV11MIXP10), the virus acquired high-level TPV resistance and replicated at TPV concentrations up to 15 µM. The IC50 of TPV against wild-type clinical strain HIV104pre was 0.16 µM, while against multi-PI-resistant strains, IC50 values ranged from 0.066 µM to 0.41 µM. [1] Introduction of I54V/V82T into cHIVB (cHIVBI54V/V82T) compromised TPV's dimerization inhibition and conferred significant TPV resistance, with an IC50 of 3.2 µM (12-fold increase). [1] L24M substitution in cHIVC conferred moderate resistance to TPV, with IC50 increasing from 0.9 µM to 2.2 µM. [1] L33I substitution in cHIVB did not significantly alter TPV susceptibility, but in combination with I54V or I54V/V82T, it increased resistance. [1] |
| ln Vivo |
To increase the bioavailability of dipranavir (PNU-140690), it is necessary to combine it with low-dose ritonavir (RTV) when given orally twice a day. The abundance of Tipranavir (PNU-140690) in the liver, spleen, and eyes is considerably higher in Tipranavir/r-cotreated mice than in Tipranavir-treated animals. In the Tipranavir-alone group, 31 and 38% of the serum and liver, respectively, are made up of metabolites of tipranavir (PNU-140690). In the serum and liver of mice cotreated with tipranavir (PNU-140690) and tipranavir (TPV/r), respectively, only 1 and 2% of metabolites are found. One dose of [14C]Tipranavir (PNU-140690) is given to Sprague-Dawley rats in conjunction with RTV. Feces include a lot of oxidation-related metabolites. There isn't a single metabolite that is discovered to be substantially present in urine[2]. Mice were orally administered Tipranavir (40 mg/kg or 100 mg/kg) with or without RTV (40 mg/kg). TPV and its metabolites were found in feces but not in urine. Eight TPV metabolites were identified in mouse feces, including three monohydroxylated, three desaturated, one dealkylated, and one dihydroxylated metabolites. [2] Co-administration of RTV significantly inhibited all eight TPV metabolic pathways in mice. [2] TPV tissue distribution was highest in the liver, followed by kidney, spleen, and lung. Low levels were detected in brain and eyes. RTV co-treatment increased TPV abundance in liver, spleen, and eyes. [2] |
| Enzyme Assay |
An intermolecular FRET-based HIV-1 expression system was used to assess protease dimerization inhibition. CFP- and YFP-tagged HIV-1 protease monomers were co-expressed in COS7 cells. FRET signals were measured using confocal microscopy. A CFPA/B ratio >1 indicates dimerization, while <1 indicates inhibition. [1] In the presence of 1 µM and 10 µM TPV, the CFPA/B ratios for wild-type protease were 0.85 and 0.64, respectively, indicating dimerization inhibition. [1] Substitutions such as L24M, L33I, L33F, and E34D were shown to compromise TPV's dimerization inhibition activity. [1] |
| Cell Assay |
Drug susceptibility assays were performed using PHA-stimulated PBMCs or MT-4 cells. Cells were infected with HIV-1 isolates (50 TCID50) and cultured in the presence of serial dilutions of TPV. After 7 days, p24 Gag protein production was measured using a chemiluminescent enzyme immunoassay. IC50 values were calculated based on p24 inhibition. [1] Selection experiments were conducted by passaging HIV-1 in MT-4 cells with escalating TPV concentrations (starting from 0.4 µM up to 15 µM). Viral replication was monitored by p24 production, and proviral DNA was sequenced to identify mutations. [1] |
| Animal Protocol |
Mice[2] All mice (2-4 months old) are maintained under a standard 12-h dark and 12-h light cycle with water and chow provided ad libitum. For metabolomic analysis, Tipranavir (40 mg/kg) is administered via ball-tipped gavage needles, and the mice are housed in separate metabolic cages for 18 h. Urine and feces samples are collected and stored at 20°C for further analysis. For tissue distribution and inhibition studies, three groups of mice are used and are orally treated with Tipranavir (100 mg/kg), RTV (40 mg/kg), and Tipranavir/r (100 mg/kg Tipranavir and 40 mg/kg RTV), respectively. Tissues including the liver, brain, lung, kidney, spleen, and eyes are collected 30 min after treatment and stored at 20°C for further analysis. Mice (2–4 months old) were orally administered Tipranavir at 40 mg/kg (for metabolomic analysis) or 100 mg/kg (for tissue distribution study) using ball-tipped gavage needles. [2] For the inhibition study, mice were treated with TPV (100 mg/kg), RTV (40 mg/kg), or TPV/r (100 mg/kg TPV + 40 mg/kg RTV). [2] Animals were housed in metabolic cages for 18 hours post-dose for urine and feces collection. Tissues (liver, brain, lung, kidney, spleen, eyes) were collected 30 minutes after treatment. [2] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Absorption is limited, although no absolute quantification of absorption is available. Tipranavir is extensively bound to plasma proteins (>99.9%). It binds to both human serum albumin and a-1-acid glycoprotein. The mean fraction of tipranavir (dosed without ritonavir) unbound in plasma was similar in clinical samples from healthy volunteers and HIV-1 positive patients. Total plasma tipranavir concentrations for these samples ranged from 9 to 82 uM. The unbound fraction of tipranavir appeared to be independent of total drug concentration over this concentration range. Administration of (14)C-tipranavir to subjects (n=8) that received Aptivus/ritonavir 500/200 mg dosed to steady-state demonstrated that most radioactivity (median 82.3%) was excreted in feces, while only a median of 4.4% of the radioactive dose administered was recovered in urine. In addition, most radioactivity (56%) was excreted between 24 and 96 hours after dosing. The effective mean elimination half-life of tipranavir/ritonavir in healthy volunteers (n=67) and HIV-1 infected adult patients (n=120) was approximately 4.8 and 6.0 hours, respectively, at steady state following a dose of 500/200 mg twice daily with a light meal. The pharmacokinetic and metabolite profiles of the antiretroviral agent tipranavir (TPV), administered with ritonavir (RTV), in nine healthy male volunteers were characterized. Subjects received 500-mg TPV capsules with 200-mg RTV capsules twice daily for 6 days. They then received a single oral dose of 551 mg of TPV containing 90 uCi of [(14)C]TPV with 200 mg of RTV on day 7, followed by twice-daily doses of unlabeled 500-mg TPV with 200 mg of RTV for up to 20 days. Blood, urine, and feces were collected for mass balance and metabolite profiling. Metabolite profiling and identification was performed using a flow scintillation analyzer in conjunction with liquid chromatography-tandem mass spectrometry. The median recovery of radioactivity was 87.1%, with 82.3% of the total recovered radioactivity excreted in the feces and less than 5% recovered from urine. Most radioactivity was excreted within 24 to 96 hr after the dose of ((14)C)TPV. Radioactivity in blood was associated primarily with plasma rather than red blood cells. Unchanged TPV accounted for 98.4 to 99.7% of plasma radioactivity. Similarly, the most common form of radioactivity excreted in feces was unchanged TPV, accounting for a mean of 79.9% of fecal radioactivity. The most abundant metabolite in feces was a hydroxyl metabolite, H-1, which accounted for 4.9% of fecal radioactivity. TPV glucuronide metabolite H-3 was the most abundant of the drug-related components in urine, corresponding to 11% of urine radioactivity. In conclusion, after the coadministration of TPV and RTV, unchanged TPV represented the primary form of circulating and excreted TPV and the primary extraction route was via the feces. The in vitro plasma protein binding of tipranavir was very high (> 99.9%) in all species including humans, with only a slight trend towards saturation over the concentration range of 10 to 100 um. Tipranavir with or without ritonavir co-administration, distributed primarily in the liver, small intestine, large intestine, kidney and lung. Tipranavir did not cross the blood-brain barrier and did not readily partitioning into red blood cells. Following intravenous dosing, tipranavir demonstrated low clearance ranging from 0.08 L/hr/kg in dogs to 1.15 l/h/kg in mice. The Vss ranged from 0.13 L/kg in dogs to 0.51 L/kg in rats. TPV was eliminated rapidly with a terminal t1/2 ranging from 0.93 hr in dogs to 5.43 hr in rats. Following oral dosing, tipranavir exhibited a mean Tmax ranging from 0.5 to 8 hr in all species. In all species a moderate or poor oral bioavailability of tipranavir was revealed, due to a lack of absorption and/or intestinal metabolism. Whereas the bioavailability in rats showed moderately levels of 28.0%, the bioavailability in dogs (6.5% and 7.7%) and also in mice (11%) and rabbits (9.9%) was minimal. Food had no significant effect on tipranavir oral bioavailability in dogs. Ritonavir co-administration studies were performed to investigate the benefit gained by the combination. However the use of different doses of ritonavir for oral and intravenous PK of tipranavir does not allow a clear comparison of tipranavir bioavailability with or without ritonavir. With ritonavir co-administration, following intravenous dosing, tipranavir demonstrated low to moderate clearance ranging from 0.0182 L/hr/kg in rats to 3.00 L/hr/kg in mice. In rats and dogs, co-administration of ritonavir resulted in a 4- to 5-fold decrease in clearance for tipranavir, which would be consistent with inhibition of drug-metabolising enzymes by ritonavir. Metabolism / Metabolites Hepatic. In vitro metabolism studies with human liver microsomes indicated that CYP 3A4 is the predominant CYP enzyme involved in tipranavir metabolism. Tipranavir (TPV) is the first nonpeptidic protease inhibitor used for the treatment of drug-resistant HIV infection. Clinically, TPV is coadministered with ritonavir (RTV) to boost blood concentrations and increase therapeutic efficacy. The mechanism of metabolism-mediated drug interactions associated with RTV-boosted TPV is not fully understood. In the current study, TPV metabolism was investigated in mice using a metabolomic approach. TPV and its metabolites were found in the feces of mice but not in the urine. Principal component analysis of the feces metabolome uncovered eight TPV metabolites, including three monohydroxylated, three desaturated, one dealkylated, and one dihydroxylated. In vitro study using human liver microsomes recapitulated five TPV metabolites, all of which were suppressed by RTV. CYP3A4 was identified as the primary enzyme contributing to the formation of four TPV metabolites (metabolites II, IV, V, and VI), including an unusual dealkylated product arising from carbon-carbon bond cleavage. Multiple cytochromes P450 (2C19, 2D6, and 3A4) contributed to the formation of a monohydroxylated metabolite (metabolite III). In vivo, RTV cotreatment significantly inhibited eight TPV metabolic pathways. In summary, metabolomic analysis revealed two known and six novel TPV metabolites in mice, all of which were suppressed by RTV. The current study provides solid evidence that the RTV-mediated boosting of TPV is due to the modulation of P450-dependent metabolism. The pharmacokinetic and metabolite profiles of the antiretroviral agent tipranavir (TPV), administered with ritonavir (RTV), in nine healthy male volunteers were characterized. Subjects received 500-mg TPV capsules with 200-mg RTV capsules twice daily for 6 days. They then received a single oral dose of 551 mg of TPV containing 90 uCi of [(14)C]TPV with 200 mg of RTV on day 7, followed by twice-daily doses of unlabeled 500-mg TPV with 200 mg of RTV for up to 20 days. ... The most abundant metabolite in feces was a hydroxyl metabolite, H-1, which accounted for 4.9% of fecal radioactivity. TPV glucuronide metabolite H-3 was the most abundant of the drug-related components in urine, corresponding to 11% of urine radioactivity. ... In vitro metabolism studies indicated that CYP3A4 is the predominant CYP isoform involved in tipranavir metabolism in humans. CYP3A isozyme was also identified in rat as the predominant CYP isoform involved in tipranavir metabolism. Studies in rats and humans dosed by tipranavir co-administered with ritonavir were conducted to assess metabolites. The unchanged tipranavir was the predominant form in plasma (>85.7%). Unchanged tipranavir was also the major form excreted in feces and urine. Combined levels of excreted metabolites in feces and urine accounted for approximately 4.8% and 7.4% in male and female rats. Only small amounts of a glucuronide were observed in faeces. For more Metabolism/Metabolites (Complete) data for Tipranavir (6 total), please visit the HSDB record page. Biological Half-Life 5-6 hours Tipranavir and its metabolites were excreted primarily in feces, not in urine. [2] In mice, TPV tissue distribution was highest in the liver. Co-administration with RTV increased TPV levels in liver, spleen, and eyes. [2] In serum and liver of TPV-alone mice, metabolites accounted for 31% and 38% of total TPV-related material, respectively. In TPV/r-treated mice, metabolites accounted for only 1–2%. [2] The study suggests that TPV metabolism is mediated mainly by CYP3A4, with contributions from CYP2C19 and CYP2D6 for certain metabolites. [2] |
| Toxicity/Toxicokinetics |
Hepatotoxicity Some degree of serum aminotransferase elevations occur in a high proportion of patients taking tipranavir containing antiretroviral regimens. Moderate-to-severe elevations in serum aminotransferase levels (>5 times the upper limit of normal) are found in 3% to 10% of patients, although rates may be higher in patients with HIV-HCV coinfection. These elevations are usually asymptomatic and self-limited and can resolve even with continuation of the medication. Clinically apparent liver injury from tipranavir is rare, and the clinical pattern of liver injury, latency and recovery have not been well defined. Several protease inhibitors have been associated with acute liver injury arising 1 to 8 weeks after onset, with variable patterns of liver enzyme elevation, from hepatocellular to cholestatic. Immunoallergic features (rash, fever, eosinophilia) are uncommon, as is autoantibody formation. The acute liver injury due to tipranavir is usually self-limited, but it can be severe, and isolated cases of acute liver failure have been reported to the sponsor. In HBV or HCV coinfected patients, some instances appear to be due to exacerbation of the underlying chronic liver disease, perhaps as a result of sudden immune reconstitution. Tipranavir therapy has not been clearly linked to lactic acidosis and acute fatty liver that is reported in association with several nucleoside analogue reverse transcriptase inhibitors. Thus, tipranavir is associated with a high rate of serum enzyme elevations which is generally higher than with other protease inhibitors, for which reason it is considered a second-line HIV protease inhibitor. Likelihood score: E (unproven but suspected cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No published information is available on the use of tipranavir during breastfeeding. Tipranavir is not recommended during breastfeeding. Achieving and maintaining viral suppression with antiretroviral therapy decreases breastfeeding transmission risk to less than 1%, but not zero. Individuals with HIV who are on antiretroviral therapy with a sustained undetectable viral load and who choose to breastfeed should be supported in this decision. If a viral load is not suppressed, banked pasteurized donor milk or formula is recommended. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Gynecomastia has been reported among men receiving highly active antiretroviral therapy. Gynecomastia is unilateral initially, but progresses to bilateral in about half of cases. No alterations in serum prolactin were noted and spontaneous resolution usually occurred within one year, even with continuation of the regimen. Some case reports and in vitro studies have suggested that protease inhibitors might cause hyperprolactinemia and galactorrhea in some male patients, although this has been disputed. The relevance of these findings to nursing mothers is not known. The prolactin level in a mother with established lactation may not affect her ability to breastfeed. Protein Binding Extensive (> 99.9%), to both human serum albumin and α-1-acid glycoprotein. Interactions Pharmacokinetic interaction with fluconazole (increased tipranavir concentrations; no change in fluconazole concentrations and AUC). If ritonavir-boosted tipranavir and fluconazole are used concomitantly, fluconazole dosage does not need to be adjusted but fluconazole dosage exceeding 200 mg daily is not recommended. If high fluconazole dosage is indicated, an alternative HIV PI or antiretroviral agent from another class should be considered. Possible pharmacokinetic interaction with carbamazepine, phenobarbital, or phenytoin (decreased tipranavir concentrations and possible decreased antiretroviral efficacy; altered carbamazepine concentrations). If used with carbamazepine or phenytoin, some experts suggest that anticonvulsant and tipranavir concentrations be monitored; alternatively, use of another anticonvulsant can be considered. Possible pharmacokinetic interaction with valproic acid (decreased plasma concentrations of valproic acid); possibility that the anticonvulsant may be less effective. Possible pharmacokinetic interaction with warfarin (altered warfarin concentrations). International normalized ratio (INR) should be monitored if warfarin is used concomitantly with ritonavir-boosted tipranavir, especially when initiating or discontinuing the antiretroviral agents; warfarin dosage should be adjusted as needed. Concomitant use of ritonavir-boosted tipranavir and an anticoagulant may increase the risk for bleeding;1 the drugs should be used concomitantly with caution. Possible pharmacokinetic interactions with amiodarone, bepridil (no longer commercially available in the US), flecainide, propafenone, or quinidine (increased plasma concentrations of the antiarrhythmic agent). Potential for serious and/or life-threatening adverse effects (e.g., cardiac arrhythmias). Concomitant use with ritonavir-boosted tipranavir is contraindicated. For more Interactions (Complete) data for Tipranavir (35 total), please visit the HSDB record page. The literature mentions that TPV/r combination therapy has been associated with intracranial hemorrhage and hepatotoxicity. More than 10 cases of intracranial hemorrhage and 12 cases of liver-associated deaths were reported. [2] TPV is highly concentrated in the liver, especially when co-administered with RTV. Disruption of biliary function may increase the risk of TPV-induced liver injury. [2] |
| References |
[1]. Loss of the protease dimerization inhibition activity of tipranavir (TPV) and its association with the acquisition of resistance to TPV by HIV-1. J Virol. 2012 Dec;86(24):13384-96. [2]. Metabolism-mediated drug interactions associated with ritonavir-boosted tipranavir in mice. Drug Metab Dispos. 2010 May;38(5):871-8. [3]. Bardoxolone and bardoxolone methyl, two Nrf2 activators in clinical trials, inhibit SARS-CoV-2 replication and its 3C-like protease. Signal Transduct Target Ther. 2021 May 29;6(1):212. |
| Additional Infomation |
Therapeutic Uses Anti-HIV Agents Tipranavir with low-dose ritonavir (ritonavir-boosted tipranavir) is used in conjunction with other antiretroviral agents for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults, adolescents, and pediatric patients 2 years of age and older with evidence of viral replication who are antiretroviral-experienced and infected with HIV-1 strains resistant to multiple HIV protease inhibitors (PIs). /Included in US product labeling/ Drug Warnings /BOXED WARNING/ WARNING: HEPATOTOXICITY and INTRACRANIAL HEMORRHAGE. Hepatotoxicity: Clinical hepatitis and hepatic decompensation, including some fatalities, have been reported. Extra vigilance is warranted in patients with chronic hepatitis B or hepatitis C co-infection, as these patients have an increased risk of hepatotoxicity. Intracranial Hemorrhage: Both fatal and non-fatal intracranial hemorrhage have been reported. New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus and hyperglycemia have been reported during post-marketing surveillance in HIV-1 infected patients receiving protease inhibitor therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued protease inhibitor therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and a causal relationship between protease inhibitor therapy and these events has not been established. Aptivus should be used with caution in patients with a known sulfonamide allergy. Tipranavir contains a sulfonamide moiety. The potential for cross-sensitivity between drugs in the sulfonamide class and Aptivus is unknown. Rash, including maculopapular rash, urticarial rash, and possible photosensitivity reaction, has been reported in patients receiving ritonavir-boosted tipranavir. Rash occurred in 10% of women, 8% of men, and 21% of children receiving ritonavir-boosted tipranavir in clinical studies. The median time to onset of rash was 53 days and the median duration of rash was 22 days in adults. Rash accompanied by joint pain or stiffness, throat tightness, or generalized pruritus also has been reported. Discontinue tipranavir if severe rash develops. For more Drug Warnings (Complete) data for Tipranavir (16 total), please visit the HSDB record page. Pharmacodynamics Tipranavir is a non-peptidic protease inhibitor (PI) of HIV. Protease inhibitors block the part of HIV called protease. HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Nelfinavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. Protease inhibitors are almost always used in combination with at least two other anti-HIV drugs. Tipranavir is a non-peptidic protease inhibitor used in combination antiretroviral therapy for HIV-1 infection, particularly in patients with multi-PI-resistant variants. [1] TPV inhibits both the enzymatic activity and dimerization of HIV-1 protease. Its dimerization inhibition activity is compromised by single or double amino acid substitutions (e.g., L24M, L33I/F, E34D, I54V, V82T), which may explain its relatively low genetic barrier to resistance compared to darunavir. [1] TPV-selected HIV-1 variants remained susceptible to darunavir, with an IC50 of 0.034 µM against HIV11MIXP10. [1] |
Solubility Data
| Solubility (In Vitro) |
DMSO : ~200 mg/mL (~331.86 mM) Ethanol :≥ 50 mg/mL (~82.97 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: 5 mg/mL (8.30 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 50.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 5 mg/mL (8.30 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 50.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 3: 2.5 mg/mL (4.15 mM) in 10% EtOH + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear EtOH stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 4: 2.5 mg/mL (4.15 mM) in 10% EtOH + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear EtOH stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix well. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 5: ≥ 2.5 mg/mL (4.15 mM) (saturation unknown) in 10% EtOH + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear EtOH stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 6: 2.5 mg/mL (4.15 mM) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.6593 mL | 8.2966 mL | 16.5931 mL | |
| 5 mM | 0.3319 mL | 1.6593 mL | 3.3186 mL | |
| 10 mM | 0.1659 mL | 0.8297 mL | 1.6593 mL |