Physicochemical Properties
| Molecular Formula | C7H9O6P2SCL |
| Molecular Weight | 318.60836 |
| Exact Mass | 317.928 |
| CAS # | 89987-06-4 |
| Related CAS # | Tiludronate disodium;149845-07-8;Tiludronate disodium hemihydrate;155453-10-4 |
| PubChem CID | 60937 |
| Appearance | Typically exists as solid at room temperature |
| Density | 1.82g/cm3 |
| Boiling Point | 600.7ºC at 760 mmHg |
| Flash Point | 317.1ºC |
| Index of Refraction | 1.652 |
| LogP | 2.071 |
| Hydrogen Bond Donor Count | 4 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 17 |
| Complexity | 324 |
| Defined Atom Stereocenter Count | 0 |
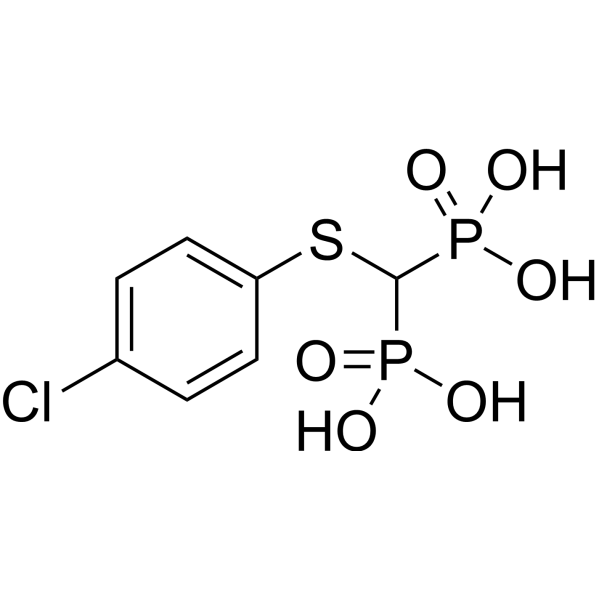
| SMILES | ClC1C=CC(SC(P(=O)(O)O)P(=O)(O)O)=CC=1 |
| InChi Key | DKJJVAGXPKPDRL-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C7H9ClO6P2S/c8-5-1-3-6(4-2-5)17-7(15(9,10)11)16(12,13)14/h1-4,7H,(H2,9,10,11)(H2,12,13,14) |
| Chemical Name | [(4-chlorophenyl)sulfanyl-phosphonomethyl]phosphonic acid |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| ln Vitro | In nephrogenic vesicles (IC50=1.1 mM) and osteoclast-derived vesicles (IC50=466 nM), tiludronate inhibits proton transport five times more potently than it does in other vesicles. Moreover, tiludronate efficiently suppresses the activity of pure yeast V-ATPase and inhibits proton transport in yeast microsomal preparations (IC50=3.5mM). Tiludronate quickly, pH-dependently, and reversibly inhibits osteoclast V-ATPase-mediated proton transport [3]. |
| ln Vivo | Tiludronate inhibits bone resorption in a dose-dependent manner. Mature osteoclasts can be affected by tiludronate through decreased proton secretion into the resorption space and increased osteoclast separation from the bone matrix. Several models of osteoporosis have also been used to test teludronate. Tiladronate sodium (5–200 mg/kg; oral) prevented the loss of bone mass in a male ovariectomized rat model. This was measured chemically by evaluating the calcium and phosphate content or physically by assessing bone weight and density[3]. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion A single 400mg dose of tiludronic acid reaches a Cmax of 3.35±1.07mg/L, with a Tmax of 1.7—0.9h, and and AUC of 27.2±9.0mg\*h/L. Tiludronic acid has an oral bioavailability of 2-11% with an average of 6%. Tiludronic acid is 60% eliminated in the urine as the unchanged parent drug. The volume of distribution of tiludronic acid is estimated to be between 30L and 60L. Due to the unknown clearance rate from bone, this may underestimate the true volume of distribution. Tiludronic acid has a renal clearance of 0.68L/h in healthy subjects and 0.47L/h in subjects with Paget's disease. Approximately 50% of tilurdronic acid binds to bone but the rate of clearance from the bone is unknown. Metabolism / Metabolites Tiludronic acid is not metabolized _in vitro_ in human liver microsomes. Biological Half-Life The mean plasma elimination half-life is 150 hours, though the elimination rate from bone is unknown. The terminal phase half life is approximately 40h after a single IV dose of 10-30mg. |
| Toxicity/Toxicokinetics |
Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Because no information is available on the use of tiludronate during breastfeeding, an alternate drug may be preferred, especially while nursing a newborn or preterm infant. However, absorption of tiludronate by a breastfed infant is unlikely. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Tiludronic acid is approximately 90% protein bound in serum. It is mostly bound to albumin. |
| References |
[1]. Prevention of postmenopausal bone loss by tiludronate. Lancet. 1989;2(8678-8679):1469-1471. [2]. Effects of local administration of tiludronic acid on experimental periodontitis in diabetic rats. J Periodontol. 2018;89(1):105-116. [3]. Tiludronate: bone pharmacology and safety. Bone. 1995;17(5 Suppl):473S-477S. [4]. The bisphosphonate tiludronate is a potent inhibitor of the osteoclast vacuolar H(+)-ATPase. J Bone Miner Res. 1996;11(10):1498-1507. |
| Additional Infomation |
Tiludronic acid is an organochlorine compound. Tiludronate, or (4-chlorophenyl)thio-methylene-1,1-bisphosphonate, is a first generation bisphosphonate similar to [etidronic acid] and [clodronic acid]. These drugs were developed to mimic the action of pyrophosphate, a regulator of calcification and decalcification. Tiludronic acid was first described in the literature in 1988 as a potential treatment for Paget's disease of bone. Tiludronic acid was granted FDA approval on 7 March 1997. Tiludronic acid is a Bisphosphonate. See also: Tiludronate Disodium (annotation moved to). Drug Indication Tiludronic acid is indicated to treat Paget's disease of bone in patients with serum alkaline phosphatase levels ≥2 times the upper limit of normal, with symptoms, or with risk of future complications. Mechanism of Action Bisphosphonates are taken into the bone where they bind to hydroxyapatite. Bone resorption by osteoclasts causes local acidification, releasing the bisphosphonate, which is taken into the osteoclast by fluid-phase endocytosis. Endocytic vesicles become acidified, releasing bisphosphonates into the cytosol of osteoclasts where they act. Osteoclasts mediate resorption of bone. When osteoclasts bind to bone they form podosomes, ring structures of F-actin. Tiludronate inhibits protein-tyrosine-phosphatase, which increases tyrosine phosphorylation, and disrupts podosome formation. Tiludronic acid also inhibits V-ATPases in the osteoclast, though the exact subunits are unknown, preventing F-actin from forming podosomes. Disruption of the podosomes causes osteoclasts to detach from bones, preventing bone resorption. |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.1386 mL | 15.6932 mL | 31.3863 mL | |
| 5 mM | 0.6277 mL | 3.1386 mL | 6.2773 mL | |
| 10 mM | 0.3139 mL | 1.5693 mL | 3.1386 mL |