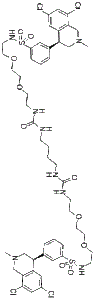
Tenapanor (formerly RDX-5791; RDX 5791; AZD-1722; AZD1722; trade name Ibsrela) is a first-in-class inhibitor of sodium-hydrogen exchanger (NHE3) approved by FDA in 2019 for the treatment of irritable bowel syndrome with constipation (IBS-C).
Physicochemical Properties
| Molecular Formula | C50H66CL4N8O10S2 |
| Molecular Weight | 1145.0486 |
| Exact Mass | 1142.309 |
| Elemental Analysis | C, 52.45; H, 5.81; Cl, 12.38; N, 9.79; O, 13.97; S, 5.60 |
| CAS # | 1234423-95-0 |
| Related CAS # | Tenapanor hydrochloride;1234365-97-9 |
| PubChem CID | 71587953 |
| Appearance | Typically exists as white to off-white solids at room temperature |
| Density | 1.3±0.1 g/cm3 |
| Index of Refraction | 1.590 |
| LogP | 4.85 |
| Hydrogen Bond Donor Count | 6 |
| Hydrogen Bond Acceptor Count | 14 |
| Rotatable Bond Count | 29 |
| Heavy Atom Count | 74 |
| Complexity | 1770 |
| Defined Atom Stereocenter Count | 2 |
| SMILES | ClC1=C([H])C(=C([H])C2=C1C([H])([H])N(C([H])([H])[H])C([H])([H])[C@@]2([H])C1C([H])=C([H])C([H])=C(C=1[H])S(N([H])C([H])([H])C([H])([H])OC([H])([H])C([H])([H])OC([H])([H])C([H])([H])N([H])C(N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])N([H])C(N([H])C([H])([H])C([H])([H])OC([H])([H])C([H])([H])OC([H])([H])C([H])([H])N([H])S(C1=C([H])C([H])=C([H])C(=C1[H])[C@@]1([H])C2C([H])=C(C([H])=C(C=2C([H])([H])N(C([H])([H])[H])C1([H])[H])Cl)Cl)(=O)=O)=O)=O)(=O)=O)Cl |
| InChi Key | DNHPDWGIXIMXSA-CXNSMIOJSA-N |
| InChi Code | InChI=1S/C50H66Cl4N8O10S2/c1-61-31-43(41-27-37(51)29-47(53)45(41)33-61)35-7-5-9-39(25-35)73(65,66)59-15-19-71-23-21-69-17-13-57-49(63)55-11-3-4-12-56-50(64)58-14-18-70-22-24-72-20-16-60-74(67,68)40-10-6-8-36(26-40)44-32-62(2)34-46-42(44)28-38(52)30-48(46)54/h5-10,25-30,43-44,59-60H,3-4,11-24,31-34H2,1-2H3,(H2,55,57,63)(H2,56,58,64)/t43-,44-/m0/s1 |
| Chemical Name | 3-((S)-6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl)-N-(26-((3-((S)-6,8-dichloro-2-methyl-1,2,3,4-tetrahydroisoquinolin-4-yl)phenyl)sulfonamido)-10,17-dioxo-3,6,21,24-tetraoxa-9,11,16,18-tetraazahexacosyl)benzenesulfonamide |
| Synonyms | RDX 5791; AZD 1722; RDX-5791; AZD1722; RDX5791; AZD-1722; Tenapanor free base;Tenapanor; 1234423-95-0; KHK7791; KHK-7791; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
IC50: 5 nM (NHE3, human), 10 nM (NHE3, rat)[1] Tenapanor exhibits human and rat NHE3 with IC50 values of 5 and 10 nM, respectively. Human intestinal transporters NHE1, NHE2, TGR5, ASBT, and Pit-1 and the sodium-dependent phosphate transporter NaPiIIb are not inhibited by tenapanor at concentrations up to 10 to 30 μM[1]. |
| ln Vitro |
Tenapanor exhibits human and rat NHE3 with IC50 values of 5 and 10 nM, respectively. Human intestinal transporters NHE1, NHE2, TGR5, ASBT, and Pit-1 and the sodium-dependent phosphate transporter NaPiIIb are not inhibited by tenapanor at concentrations up to 10 to 30 μM[1]. Tenapanor (1 μM) significantly reduced apical-to-basolateral phosphate flux in human duodenum and ileum monolayer cultures across a range of apical phosphate concentrations (1-30 mM), indicating inhibition of passive paracellular phosphate absorption. [1] Tenapanor (1 μM) increased transepithelial electrical resistance (TEER) in human duodenum and ileum monolayer cultures, consistent with a decrease in paracellular permeability. [1] Tenapanor (1 μM) reduced basolateral-to-apical phosphate flux in human duodenum monolayers, comparable to its effect on apical-to-basolateral absorption, confirming a reduction in bidirectional paracellular phosphate permeability. [1] In human ileum monolayers, tenapanor reduced phosphate absorption across a luminal pH range of 6.0 to 8.0, with a greater reduction at pH 7.0 and 8.0 compared to pH 6.0. [1] In human duodenum monolayers mounted in Ussing chambers at pH 8.0, tenapanor increased TEER and decreased paracellular permeability to sodium, chloride, and phosphate, as measured by dilution and bionic potentials. [1] In CRISPR/Cas9-generated NHE3 knockout (KO) human ileum monolayers, tenapanor (1 μM) had no effect on phosphate absorption, apical phosphate concentration, or TEER, confirming that its effects are mediated exclusively via on-target NHE3 inhibition. [1] Tenapanor (1 μM) did not affect phosphate absorption in mouse ileum monolayer cultures, a model where absorption is predominantly mediated by the active transporter NaPi2b. [1] Tenapanor (1 μM) did not induce obvious changes in the localization of tight junction proteins (zona occludens-1, occludin, claudin 7, claudin 3) in human ileum monolayers after 30, 60, or 120 minutes of treatment. [1] Inhibitors of endocytosis (Pitstop 2 and dynasore) did not block the tenapanor-induced increase in TEER in ileum monolayers. [1] In human ileum monolayers, other compounds that decrease intracellular pH (nigericin, BAM15, FCCP) also increased TEER, similar to the effect of tenapanor. [1] |
| ln Vivo |
In rats, tenapanor (0.15, 0.5 mg/kg; oral) decreases the absorption of passive paracellular phosphate [1]. Rats' decreased excretion of phosphorus in their urine is further reduced when given tenapanor (0.15 mg/kg; oral; twice daily for 11 days) [2]. In a rat intestinal (jejunum) loop model, tenapanor (10 μM) reduced radioactive phosphate absorption to a level similar to that observed under sodium-free conditions. [1] In rats, an oral bolus of tenapanor (0.5 mg/kg) reduced urinary phosphate excretion following an oral phosphate bolus across a range of concentrations (0.15-1.5 M), suggesting inhibition of intestinal phosphate absorption. [1] In rats fed diets with varying phosphate content, chronic administration of tenapanor (0.5 mg/kg for 4 days) reduced urinary phosphate excretion, indicating inhibition of a constant fraction of intestinal phosphate absorption. [1] In an enteropooling study in rats, tenapanor (0.15 mg/kg) reduced urinary phosphate and sodium excretion after a high-phosphate meal and increased the delivery (mass) and concentration of sodium and phosphate to the cecum, confirming decreased small intestinal absorption. Tenapanor increased luminal water volume and sodium concentration but did not significantly affect cecal concentrations of potassium, calcium, or magnesium. Cecal chloride concentration increased modestly. [1] In healthy rats dosed with tenapanor (0.5 mg/kg) for 14 days, urinary sodium and phosphate excretion were decreased, while urinary chloride and potassium excretion were unaffected. Plasma sodium and phosphate concentrations were unchanged. Renal clearance of phosphate and sodium was decreased. Tenapanor had minimal effect on phosphate-regulating hormones (FGF-23, parathyroid hormone, vitamin D) in healthy rats on a regular phosphate diet. [1] RNA-seq analysis in rats treated with tenapanor for 14 days showed increased NHE3 mRNA expression in the jejunum, ileum, and proximal colon, and increased epithelial sodium channel γ subunit (ENaCγ) expression in the distal colon. Expression of chloride transporters/exchangers (SLC26A3, SLC26A6, CFTR) was unchanged. [1] In rats, tenapanor (0.5 mg/kg for 14 days) modestly but significantly decreased NaPi2b mRNA expression (~30% reduction) in the distal jejunum and ileum. Immunohistochemistry confirmed a modest decrease in NaPi2b protein staining intensity in the jejunum. [1] Brush border membrane vesicles (BBMVs) isolated from the duodenum and jejunum of tenapanor-treated rats showed little effect on sodium-dependent phosphate uptake or sodium-dependent glucose absorption. [1] In an in vivo ileum loop model in mice, tenapanor (10 μM) produced a small, non-significant decrease in phosphate absorption in both wild-type and NaPi2b knockout mice. [1] In rats, tenapanor (0.5 and 10 mg/kg) inhibited absorption of radioactive phosphate but had no effect on the absorption of radioactive mannitol, a marker of paracellular macromolecule absorption. [1] In rats, tenapanor (0.15 mg/kg) did not affect the absorption of dietary glucose from the small intestine after a standardized meal. [1] In a phase 1 study in healthy human volunteers, tenapanor (15 mg twice daily for 4 days) significantly increased mean daily stool phosphorus excretion and significantly decreased mean daily urinary phosphorus and sodium excretion. Urinary potassium excretion was unaffected. [1] |
| Enzyme Assay |
The potency of tenapanor against NHE3 was assessed in human and mouse ileum monolayer cultures by measuring its inhibition of apical acid secretion. Apical media acidification, resulting from proton secretion coupled to sodium absorption by NHE3, was monitored using the color change of pH-sensitive phenol red or quantitatively using the fluorescent pH indicator dye BCECF-AM. For the quantitative assay, a decrease in the fluorescence emission ratio (excitation 440 nm/490 nm, emission 535 nm) reflects a decrease in pH. Concentration-response studies were performed to determine the IC50 value. [1] The effect of tenapanor on NHE3-mediated recovery of intracellular pH (pHi) after an acid load was measured in human duodenum and ileum monolayer cultures. Cells were loaded with the pH-sensitive dye BCECF-AM. Intracellular acidification was induced by incubation in sodium-free media. Recovery was initiated by adding sodium-containing media at varying luminal pH levels (e.g., 6.0, 7.0) in the presence or absence of tenapanor. The rate of pHi recovery, reflecting NHE3 activity, was monitored fluorometrically. [1] |
| Cell Assay |
Tenapanor inhibits paracellular phosphate flux in an intestinal epithelial cellular model Intestinal epithelial stem cells from human or mouse gastrointestinal biopsies cultured as monolayers allow for monitoring of ion transport across the intestinal epithelium. The enteroid monolayer contains the diversity of intestinal epithelial cell lineages, models the specific gene expression patterns of each individual intestinal segment, expresses the appropriate endogenous ion transporters (for example, NHE3 and NaPi2b) in a segment-specific manner, polarizes to form tight junctions with segment-specific expression of claudins and other tight junction proteins, and generates the expected negative luminal electrical potential observed in vivo. The differentiated enteroid monolayer therefore enables the study of transcellular and paracellular phosphate absorption[1]. Intestinal epithelial stem cell monolayer culture model[1]. Intestinal epithelial stem cell monolayers were cultured and differentiated on Transwells as described in detail by Kozuka et al.. Human biopsies from which stem cells were sourced were obtained from visibly healthy tissue from male donors according to a protocol approved by the Copernicus Group Institutional Review Board. Experiments were initiated in each differentiated monolayer culture well by washing the apical and basolateral side twice with fresh supplemented basal media and phosphate-free Dulbecco’s modified Eagle’s medium. Compounds were dosed only on the apical side of the monolayer, as detailed in the text; DMSO at an equivalent concentration was used as the control. Phosphate concentration and pH were manipulated as described in the text. Apical and basolateral ion concentrations were measured by ion chromatography, pH was measured using a pH meter, and TEER values were recorded using a volt/ohm meter. TEER results are reported as normalized to baseline. Absolute baseline TEER values are reported in table S1. Human and mouse intestinal epithelial stem cell-derived enteroid monolayers were cultured and differentiated on Transwell inserts. These monolayers model intestinal ion transport physiology, express endogenous transporters (NHE3, NaPi2b) in a segment-specific manner, form tight junctions, and generate a luminal-negative electrical potential. [1] For phosphate flux experiments, monolayers were washed and incubated with media containing defined phosphate concentrations in the apical and basolateral compartments. Tenapanor or vehicle was added to the apical side. After incubation (e.g., 4 hours or overnight), phosphate concentrations in the apical and basolateral media were measured by ion chromatography. Phosphate flux was calculated. Transepithelial electrical resistance (TEER) was measured using a volt/ohm meter. [1] To assess paracellular ion permeability, human duodenum monolayers or mouse jejunum strips were mounted in Ussing chambers. Sodium chloride dilution potentials and phosphate bionic potentials were measured under short-circuit conditions to calculate paracellular permeability to sodium (pNa+), chloride (pCl-), and phosphate (pPO43-). Tenapanor or vehicle was added to the mucosal side. [1] Intracellular pH (pHi) was measured using the pH-sensitive fluorescent dye BCECF-AM. Cells were loaded with the dye, and fluorescence was measured with dual excitation (440 nm and 490 nm) and emission at 535 nm. The ratio of fluorescence (490/440) is pH-dependent. Changes in pHi were monitored after manipulations such as changing apical media pH or adding tenapanor. [1] CRISPR/Cas9-mediated gene editing was used to generate NHE3 knockout human ileum epithelial stem cell clones. Knockout was confirmed by DNA sequencing, Western blot for NHE3 protein, and functional assays (lack of apical media acidification, absence of pHi recovery after acid load). These knockout monolayers were used to test the specificity of tenapanor's effects. [1] The role of endocytosis in the tenapanor-induced TEER increase was investigated by pretreating ileum monolayers with inhibitors of clathrin-mediated endocytosis (Pitstop 2) or dynamin (dynasore). Their efficacy was confirmed by their ability to prevent carbachol-induced NHE3 internalization. TEER was then measured after tenapanor addition. [1] The effect of other intracellular acidifying agents on TEER was tested. Ileum monolayers were treated with the ionophore nigericin or mitochondrial uncouplers (BAM15, FCCP), which decrease pHi. TEER was measured and compared to control. [1] The localization of tight junction proteins (zona occludens-1, occludin, claudin 7, claudin 3) was assessed by immunocytochemistry in human ileum monolayers treated with tenapanor or vehicle for 30, 60, and 120 minutes. [1] The effect of a NaPi2b inhibitor (NTX-9066) was tested in mouse ileum monolayers, where phosphate absorption is predominantly transcellular and NaPi2b-mediated. Monolayers were incubated with the inhibitor, tenapanor, or vehicle for various durations (4 hours, 2 days, 3 days), and apical phosphate concentration was measured. [1] |
| Animal Protocol |
Animal/Disease Models: Rats (intestinal loop model)[1] Doses: 0.15, 0.5 mg/kg Route of Administration: Po Experimental Results: decreased passive paracellular phosphate absorption by decreased urinary phosphate and sodium excretion after the high-phosphate meal and increased sodium and phosphate delivery to the cecum. Animal/Disease Models: 8 weeks, 250 g male Sprague–Dawley rats[2] Doses: 0.15 mg/kg in combination with sevelamer (0%, 0.75%, 1.5%, and 3% (wt/wt)) Route of Administration: po (oral gavage); twice-daily for 11 days Experimental Results: Dramatically augmented the reduction in urinary phosphorus excretion. Rat intestinal loop model: Rats were anesthetized. A segment of jejunum was isolated, and the lumen was rinsed. A buffer containing radioactive phosphate (33P) with or without tenapanor (10 μM) or with sodium-free buffer was injected into the loop. The loop was returned to the abdomen for a specified time. Phosphate absorption was determined by measuring the disappearance of radioactivity from the loop and/or its appearance in plasma. [1] Oral phosphate bolus and urinary excretion in rats: Rats were pretreated with tenapanor (0.5 mg/kg) or vehicle by oral gavage. Subsequently, an oral bolus of phosphate solution at varying concentrations (0.15-1.5 M) was administered. Urine was collected for 4 hours, and phosphate excretion was measured. [1] Chronic dietary phosphate study in rats: Rats were fed diets with different phosphate contents. After baseline urine collection, rats were treated with tenapanor (0.5 mg/kg) or vehicle orally twice daily for 4 days. Urine was collected during treatment for phosphate excretion analysis. [1] Enteropooling study in rats: Rats were trained to consume a standardized high-phosphate (1.2%) meal within a short period. On the study day, rats were treated with tenapanor (0.15 mg/kg) or vehicle before the meal. At defined time points after meal initiation, rats were euthanized, and the cecum was removed. Cecal contents were weighed, and the concentrations of ions (sodium, phosphate, potassium, chloride, calcium, magnesium) were measured by ion chromatography. Urine was also collected for ion excretion analysis. [1] 14-day repeat dose study in rats: Healthy rats were dosed orally with tenapanor (0.5 mg/kg) or vehicle twice daily for 14 days. Urine was collected over 24-hour periods at various time points for measurement of sodium, phosphate, chloride, and potassium excretion. Blood was collected at the end for plasma ion and hormone (FGF-23, PTH, vitamin D) analysis. Renal clearance was calculated. Gastrointestinal tissues were collected for RNA-seq analysis and NaPi2b mRNA expression measurement by qPCR. [1] NaPi2b expression and BBMV study in rats: In a separate study, rats were treated with tenapanor or vehicle for a period. Jejunum tissue was collected for immunohistochemistry staining of NaPi2b protein. For BBMV preparation, duodenum and jejunum were collected from treated rats. BBMVs were isolated by a Mg2+ precipitation method. Uptake of radioactive phosphate or glucose into BBMVs was measured in the presence or absence of sodium. [1] Mouse ileum loop model: Wild-type and NaPi2b knockout mice were used. Under anesthesia, an ileal loop was prepared and injected with a buffer containing radioactive phosphate and tenapanor (10 μM) or vehicle. Phosphate absorption was determined after a set incubation period. [1] Mannitol and glucose absorption studies in rats: For mannitol absorption, rats received an oral dose of tenapanor (0.5 or 10 mg/kg) or vehicle, followed by an oral dose containing 3H-mannitol and 33P-phosphate. Blood was sampled over time to measure radioactivity. For glucose absorption, rats pretreated with tenapanor (0.15 mg/kg) or vehicle consumed a standardized meal over 4 hours. At specified times, rats were euthanized, and the entire small intestine was removed. Luminal glucose content was measured. [1] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Tenapanor undergoes very minimal systemic absorption following oral administration. During clinical trials, plasma concentrations were below the limit of quantitation (i.e. less than 0.5 ng/mL) in the majority of samples from healthy subjects - for this reason, typical pharmacokinetic values related to absorption such as AUC and Cmax were unable to be ascertained. The effects of tenapanor are greatest when administered 5 to 10 minutes before meals. Following administration of a radio labeled dose of tenapanor, 70% of the radioactivity was excreted in the feces within 120 hours of administration and 79% within 240 hours. Approximately 65% of the total dose is excreted as unchanged parent drug within 144 hours of administration. Only 9% of the administered dose was found in the urine, existing primarily as metabolites. Tenapanor's M1 metabolite is excreted unchanged in the urine and accounts for approximately 1.5% of the total dose within 144 hours of administration. Metabolism / Metabolites The majority of tenapanor's metabolism to its primary metabolite, M1, is catalyzed via CYP3A4/5. Exposure of tenapanor to hepatic CYP enzymes is likely limited due to its minimal systemic absorption, so its metabolism may be due to intestinal CYP enzyme activity. The M1 metabolite of tenapanor is a P-glycoprotein substrate and, in contrast to its parent drug, can be detected in plasma, reaching a Cmax of approximately 15 ng/mL at steady state. It is not considered active against NHE3. Biological Half-Life Tenapanor's FDA label states that its half-life could not be determined during clinical trials due to its minimal systemic absorption resulting in plasma concentrations below the limit of quantitation (i.e. less than 0.5 ng/mL). Tenapanor is described as a "minimally absorbed" small-molecule inhibitor that acts locally in the gastrointestinal tract. [1] In healthy human volunteers, systemic drug exposure was reported to be minimal. [1] |
| Toxicity/Toxicokinetics |
Hepatotoxicity When given orally, tenapanor has minimal systemic absorption and has not been associated with elevations in serum enzymes or bilirubin or with instances of clinically apparent liver injury. Since approval and general availability of tenapanor, there have been no published reports of liver injury attributed to its use. Likelihood score: E (unlikely cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Tenapanor is essentially non-absorbed systemically, with undetectable plasma concentrations following oral administration. The minimal systemic absorption of tenapanor will not result in a clinically relevant exposure to breastfed infants. No special precautions are necessary. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Both tenapanor and its principle metabolite, M1, are highly plasma protein bound at approximately 99% and 97%, respectively. The specific proteins to which tenapanor and its metabolite binds have yet to be elucidated. The study mentions that in healthy volunteers, treatment with tenapanor did not affect serum bicarbonate or urinary pH, suggesting no perturbation of systemic acid-base balance. [1] |
| References |
[1]. Inhibition of sodium/hydrogen exchanger 3 in the gastrointestinal tract by tenapanor reduces paracellular phosphate permeability. Sci Transl Med. 2018 Aug 29;10(456):eaam6474. [2]. Combination treatment with tenapanor and sevelamer synergistically reduces urinary phosphorus excretion in rats. Am J Physiol Renal Physiol. 2021 Jan 1;320(1):F133-F144. |
| Additional Infomation |
Tenapanor is a novel, small molecule medication approved in September 2019 for the treatment of constipation-predominant irritable bowel-syndrome (IBS-C). It was first designed and synthesized in 2012. As an inhibitor of the sodium/hydrogen exchanger isoform 3 (NHE3) transporter, it is the first and currently only medication within its class and therefore exists as a novel alternative in the treatment of IBS-C. In October 2023, tenapanor was approved for the treatment of chronic kidney disease. Tenapanor is a Sodium-Hydrogen Exchanger 3 Inhibitor. The mechanism of action of tenapanor is as a Sodium-Hydrogen Exchanger 3 Inhibitor, and Organic Anion Transporting Polypeptide 2B1 Inhibitor. Tenapanor is a small molecular inhibitor of the sodium/hydrogen ion exchanger-3 (NHE3) used to treat constipation predominant irritable bowel syndrome (IBS). Tenapanor has minimal systemic absorption and has not been associated with serum enzyme elevation during therapy nor has it been linked to cases of clinically apparent liver injury. See also: Tenapanor Hydrochloride (active moiety of). Drug Indication Tenapanor is indicated for the treatment of constipation-predominant irritable bowel syndrome (IBS-C) in adults. It is also indicated to reduce serum phosphorus in adults with chronic kidney disease (CKD) on dialysis as add-on therapy in patients who have an inadequate response to phosphate binders or who are intolerant of any dose of phosphate binder therapy. Mechanism of Action Tenapanor is a locally-acting small molecule inhibitor of the sodium/hydrogen exchanger isoform 3 (NHE3), an antiporter expressed on the apical surface of enterocytes in the small intestine and colon which is involved in sodium-fluid homeostasis. By inhibiting this antiporter tenapanor causes retention of sodium within the lumen of the intestine - this results in an osmotic gradient that draws water into the lumen and softens stool consistency. There is some evidence that tenapanor can inhibit the uptake of dietary phosphorus in the gastrointestinal tract, though the exact mechanism of this activity has yet to be elucidated. Pharmacodynamics Through the inhibition of dietary sodium absorption tenapanor causes an increase in water secretion into the intestines, thereby decreasing transit time and softening stool consistency. Tenapanor is an investigational drug for the treatment of hyperphosphatemia in patients with chronic kidney disease (CKD), particularly end-stage renal disease (ESRD) on dialysis. [1] Its primary mechanism of action is local inhibition of the sodium/hydrogen exchanger 3 (NHE3) in the gastrointestinal tract. This inhibition leads to intracellular acidification in enterocytes, which modulates tight junctions, increases transepithelial electrical resistance (TEER), and specifically reduces the paracellular permeability to phosphate. This reduces the passive paracellular absorption of dietary phosphate, which is quantitatively the major route of phosphate absorption at typical luminal concentrations. [1] Tenapanor also modestly decreases the expression of the active intestinal phosphate transporter NaPi2b, preventing compensatory increases in transcellular phosphate uptake. It does not directly inhibit NaPi2b activity. [1] The effect of tenapanor on ion absorption appears specific to sodium and phosphate. It does not significantly affect the absorption of potassium, calcium, magnesium, chloride (overall balance), glucose, or macromolecules like mannitol under physiological conditions. [1] Tenapanor retains its ability to inhibit phosphate absorption even at high luminal phosphate concentrations, unlike phosphate binders. This may allow for a less restricted diet in patients. [1] Clinical studies cited in the literature indicate that tenapanor significantly reduces serum phosphate and fibroblast growth factor 23 (FGF-23) in ESRD patients with hyperphosphatemia receiving dialysis. [1] A limitation noted in the study is that the molecular identity of the paracellular phosphate pore affected by tenapanor remains unknown. [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~50 mg/mL (~43.67 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (2.18 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (2.18 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (2.18 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 2.5 mg/mL (2.18 mM) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.8733 mL | 4.3666 mL | 8.7332 mL | |
| 5 mM | 0.1747 mL | 0.8733 mL | 1.7466 mL | |
| 10 mM | 0.0873 mL | 0.4367 mL | 0.8733 mL |