Temsavir (formerly known as BMS626529; BMS-626529) is a novel, potent attachment inhibitor that targets HIV-1 gp120 and prevents its binding to CD4+ T cells. Temsavir is also the phosphonooxymethyl prodrug of BMS-626529 that targets HIV-1 gp120 and prevents its binding to CD4(+) T cells. BMS-626529 had half-maximal effective concentration (EC(50)) values of<10 nm, with half-maximal effective concentration values in the low pM range against the most susceptible viruses.
Physicochemical Properties
| Molecular Formula | C24H23N7O4 |
| Molecular Weight | 473.48392 |
| Exact Mass | 473.181 |
| Elemental Analysis | C, 60.88; H, 4.90; N, 20.71; O, 13.52 |
| CAS # | 701213-36-7 |
| Related CAS # | Temsavir;701213-36-7;Fostemsavir Tris;864953-39-9; 864953-29-7(free base); 864953-39-9 (tromethamine) ; 864953-31-1 (disodium); 942117-71-7 (dihydrate) |
| PubChem CID | 11317439 |
| Appearance | White to off-white solid powder. |
| Density | 1.5±0.1 g/cm3 |
| Boiling Point | 787.6±70.0 °C at 760 mmHg |
| Flash Point | 430.1±35.7 °C |
| Vapour Pressure | 0.0±2.7 mmHg at 25°C |
| Index of Refraction | 1.722 |
| LogP | -1.49 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 35 |
| Complexity | 799 |
| Defined Atom Stereocenter Count | 0 |
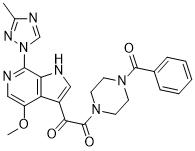
| SMILES | O=C(N1CCN(C(C2=CC=CC=C2)=O)CC1)C(C3=CNC4=C3C(OC)=CN=C4N5C=NC(C)=N5)=O |
| InChi Key | QRPZBKAMSFHVRW-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C24H23N7O4/c1-15-27-14-31(28-15)22-20-19(18(35-2)13-26-22)17(12-25-20)21(32)24(34)30-10-8-29(9-11-30)23(33)16-6-4-3-5-7-16/h3-7,12-14,25H,8-11H2,1-2H3 |
| Chemical Name | 1-(4-benzoylpiperazin-1-yl)-2-(4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-3-yl)ethane-1,2-dione |
| Synonyms | BMS-626529; BMS 626529; BMS-626529; Temsavir (BMS-626529); 1-(4-benzoylpiperazin-1-yl)-2-(4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-3-yl)ethane-1,2-dione; 4B6J53W8N3 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | HIV-1 |
| ln Vitro | Half-maximal effective concentration (EC50) values for temsavir against the majority of viral isolates are less than 10 nM. Temsavir has an average EC50 of 0.7±0.4 nM against the LAI virus. When it comes to the most susceptible virus, temsavir has an EC50 of 0.01 nM, and when it comes to the least susceptible virus, it is >2,000 nM. Temsavir's cytotoxicity profile is investigated in a variety of cell types from various human tissues. Following three or six days in culture, CC50 values of >200 μM were found in MT-2 (t lymphocytes), HEK293 (kidney), HEp-2 (larynx), HepG2 (liver), HeLa (cervix), HCT116 (colorectal), MCF-7 (breast), SK-N-MC (neuroepithelium), HOS (bone), H292 (lung), and MDBK (bovine kidney) cells.After six days in culture, PBMCs and the T-cell line PM1 have CC50 values of 192 μM and 105 μM, respectively. Temsavir demonstrates minimal cytotoxicity in cell culture, according to these findings[1]. Against a panel of clinical isolates, temsavir demonstrates a wide range of antiviral activity, with a 50% inhibitory concentration (IC50) that spans from subnanomolar levels to >0.1 µM[2]. |
| ln Vivo | The maximum median decrease in plasma HIV-1 RNA load from baseline ranged from 1.21 to 1.73 log(10) copies/mL. Plasma concentrations of BMS-626529 were not associated with an antiviral response, while low baseline inhibitory concentrations and the minimum and average steady-state BMS-626529 plasma concentrations, when adjusted by the baseline protein binding-adjusted 90% inhibitory concentration (inhibitory quotient), were linked with antiviral response. BMS-663068 was generally well tolerated. Conclusions: Administration of BMS-663068 for 8 days with or without ritonavir resulted in substantial declines in plasma HIV-1 RNA levels and was generally well tolerated. Longer-term clinical trials of BMS-663068 as part of combination antiretroviral therapy are warranted. Clinical Trials Registration.NCT01009814.[2] |
| Enzyme Assay | The binding of [3H]Temsavir or [3H]BMS-488043 to gp120 is measured using Micro BioSpin 6 columns. Binding solutions (30 μL) with serial dilutions of [3H]BMS-488043 or [3H]Temsavir, 125 mM NaCl, 50 nM gp120JRFL, and 25 mM Tris-HCl (pH 7.5) are allowed to equilibrate before being adsorbed to a MicroBioSpin 6 column. After five minutes of centrifuging the column at about 14,000 rpm, the eluent is collected, and a scintillation counter is used to measure radioactivity.Dissociative kinetics are measured by first achieving equilibrium binding with 60 nM gp120 at room temperature for 1 h, then adding a large molar excess (14-fold) of soluble CD4 protein to drive dissociation. This process is repeated with 150 nM [3H]Temsavir or 90 nM [3H]BMS-488043. The radioactivity in the eluent is quantified after aliquots are taken at the designated intervals, adsorbed to a spin column, and centrifuged. Percentage of compound bound was determined by comparing the tritium signal between parallel samples with and without the soluble CD4 challenge[1]. |
| Cell Assay | Cell viability is measured using an XTT assay, and cytotoxicity tests are conducted for up to six days in the presence of serially diluted Temsavir. Initially, 0.1×106 cells/mL is the plating density of laboratory-adapted peripheral blood mononuclear cells (PBMCs) in order to calculate CC50 values (drug concentration needed to kill 50% of cells). Cell densities in the absence of compounds usually reach 1×106 to 1.2×106/mL after 6 days[1]. |
| Animal Protocol | Fifty HIV-1-infected subjects were randomized to 1 of 5 regimen groups (600 mg BMS-663068 plus 100 mg ritonavir every 12 hours [Q12H], 1200 mg BMS-663068 plus 100 mg ritonavir every bedtime, 1200 mg BMS-663068 plus 100 mg ritonavir Q12H, 1200 mg BMS-663068 Q12H plus 100 mg ritonavir every morning, or 1200 mg BMS-663068 Q12H) for 8 days in this open-label, multiple-dose, parallel study. The study assessed the pharmacodynamics, pharmacokinetics, and safety of BMS-663068.[2] |
| ADME/Pharmacokinetics |
Absorption The absorption of temsavir is significantly limited by suboptimal dissolution and solubility following oral administration. Fostemsavir, a phosphonooxymethyl prodrug of temsavir, has improved aqueous solubility and stability under acidic conditions as compared to its parent drug - following oral administration of fostemsavir, the absolute bioavailability is approximately 26.9%. The Cmax and AUCtau following oral administration of fostemsavir 600mg twice daily was 1770 ng/mL and 12,900 ng.h/L, respectively, with a Tmax of approximately 2 hours. Co-administration of fostemsavir with a standard meal increases its AUC by approximately 10%, while co-administration with a high-fat meal increases its AUC by approximately 81%. Route of Elimination Temsavir is highly metabolized, after which it is excreted in the urine and feces as inactive metabolites. Approximately 51% of a given dose is excreted in the urine, with <2% comprising unchanged parent drug, and 33% is excreted in the feces, of which 1.1% is unchanged parent drug. Volume of Distribution The steady-state volume of distribution of temsavir following intravenous administration is approximately 29.5 L. Clearance The mean clearance and apparent clearance of temsavir, the active metabolite of fostemsavir, are 17.9 L/h and 66.4 L/h, respectively. Metabolism / Metabolites Fostemsavir is rapidly hydrolyzed to temsavir, its active metabolite, by alkaline phosphatase(s) present at the brush border membrane of the intestinal lumen. Temsavir undergoes further biotransformation to two predominant inactive metabolites: BMS-646915, a product of hydrolysis by esterases, and BMS-930644, an N-dealkylated metabolite generated via oxidation by CYP3A4. Approximately 36.1% of an administered oral dose is metabolized by esterases, 21.2% is metabolized by CYP3A4, and <1% is conjugated by UDP-glucuronosyltransferases (UGT) prior to elimination. Both temsavir and its two predominant metabolites are known to inhibit BCRP. Biological Half-Life The half-life of temsavir is approximately 11 hours. Fostemsavir is generally undetectable in plasma following oral administration. |
| Toxicity/Toxicokinetics |
Hepatotoxicity In registration clinical trials, fostemsavir was associated with alanine aminotransferase (ALT) elevations in up to 25% of patients, but levels above 5 times the upper limit of normal (ULN) arose in only 4% of subjects. Most ALT elevations were transient, asymptomatic, and did not require dose modification or discontinuation. The more marked ALT elevations were usually attributable to other conditions or complications of HIV infection. No convincing cases of fostemsavir induced liver injury were observed in preregistration trials. Since approval of fostemsavir for use as a part of a multidrug therapy of HIV, there have been no published case reports of clinically apparent liver injury attributed to its use. Interestingly, in the large preregistration trial of fostemsavir, elevations in serum aminotransferase levels were particularly noted in patients with coinfection with either hepatitis B virus (HBV) or hepatitis C virus (HCV). The deaths from liver disease in this trial appeared to be due to worsening of the coinfection during therapy. Clearly, patients with HBV or HCV coinfection should be treated for those viral infections before or concurrent with antiretroviral therapy with fostemsavir. Likelihood score: E* (unproven but suspected cause of clinically apparent liver injury). Protein Binding Temsavir is approximately 88.4% protein-bound in plasma, primarily to serum albumin. |
| References |
[1]. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrobial Agents and Chemotherapy (2012), 56(7), 3498-3507. [2]. Pharmacodynamics, safety, and pharmacokinetics of BMS-663068, an oral HIV-1 attachment inhibitor in HIV-1-infected subjects. J Infect Dis. 2012 Oct 1;206(7):1002-11. |
| Additional Infomation | BMS-663068 is the phosphonooxymethyl prodrug of BMS-626529, a novel small-molecule attachment inhibitor that targets HIV-1 gp120 and prevents its binding to CD4(+) T cells. The activity of BMS-626529 is virus dependent, due to heterogeneity within gp120. In order to better understand the anti-HIV-1 spectrum of BMS-626529 against HIV-1, in vitro activities against a wide variety of laboratory strains and clinical isolates were determined. BMS-626529 had half-maximal effective concentration (EC(50)) values of <10 nM against the vast majority of viral isolates; however, susceptibility varied by >6 log(10), with half-maximal effective concentration values in the low pM range against the most susceptible viruses. The in vitro antiviral activity of BMS-626529 was generally not associated with either tropism or subtype, with few exceptions. Measurement of the binding affinity of BMS-626529 for purified gp120 suggests that a contributory factor to its inhibitory potency may be a relatively long dissociative half-life. Finally, in two-drug combination studies, BMS-626529 demonstrated additive or synergistic interactions with antiretroviral drugs of different mechanistic classes. These results suggest that BMS-626529 should be active against the majority of HIV-1 viruses and support the continued clinical development of the compound.[1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ≥ 16.67 mg/mL (~35.21 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 1.67 mg/mL (3.53 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 16.7 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 1.67 mg/mL (3.53 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 16.7 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 1.67 mg/mL (3.53 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 16.7 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1120 mL | 10.5601 mL | 21.1202 mL | |
| 5 mM | 0.4224 mL | 2.1120 mL | 4.2240 mL | |
| 10 mM | 0.2112 mL | 1.0560 mL | 2.1120 mL |