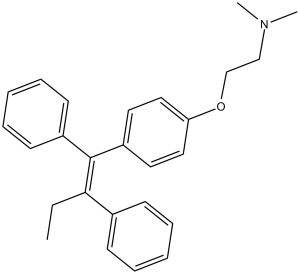
Tamoxifen (ICI-46474; NSC-180973; Nolvadex; Novaldex) is a potent and selective estrogen receptor modulator (SERM) with potential antitumor activity. It acts by competitively inhibiting estrogen binding to the ER receptors in breast tissues. In other tissues (e.g. endometrium), Tamoxifen acts as an agonist, and thus may be characterized as a selective estrogen-receptor modulator. Tamoxifen is the usual endocrine (anti-estrogen) therapy for hormone receptor-positive breast cancer in pre-menopausal women, and is also a standard in post-menopausal women although aromatase inhibitors are also frequently used in that setting [1,2]. Tamoxifen can induce gene knockout of CreER(T2) transgenic mouse [3].
Physicochemical Properties
| Molecular Formula | C26H29NO |
| Molecular Weight | 371.51 |
| Exact Mass | 371.224 |
| Elemental Analysis | C, 84.06; H, 7.87; N, 3.77; O, 4.31 |
| CAS # | 10540-29-1 |
| Related CAS # | Tamoxifen Citrate;54965-24-1;Tamoxifen (Standard);10540-29-1;Tamoxifen-d5;157698-32-3;Tamoxifen-d3;508201-30-7 |
| PubChem CID | 2733526 |
| Appearance | White to off-white solid powder |
| Density | 1.0±0.1 g/cm3 |
| Boiling Point | 482.3±33.0 °C at 760 mmHg |
| Melting Point | 97-98ºC |
| Flash Point | 140.0±27.7 °C |
| Vapour Pressure | 0.0±1.2 mmHg at 25°C |
| Index of Refraction | 1.582 |
| LogP | 7.88 |
| Hydrogen Bond Donor Count | 0 |
| Hydrogen Bond Acceptor Count | 2 |
| Rotatable Bond Count | 8 |
| Heavy Atom Count | 28 |
| Complexity | 463 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | O(C([H])([H])C([H])([H])N(C([H])([H])[H])C([H])([H])[H])C1C([H])=C([H])C(=C([H])C=1[H])/C(/C1C([H])=C([H])C([H])=C([H])C=1[H])=C(\C1C([H])=C([H])C([H])=C([H])C=1[H])/C([H])([H])C([H])([H])[H] |
| InChi Key | NKANXQFJJICGDU-QPLCGJKRSA-N |
| InChi Code | InChI=1S/C26H29NO/c1-4-25(21-11-7-5-8-12-21)26(22-13-9-6-10-14-22)23-15-17-24(18-16-23)28-20-19-27(2)3/h5-18H,4,19-20H2,1-3H3/b26-25- |
| Chemical Name | 2-[4-[(Z)-1,2-diphenylbut-1-enyl]phenoxy]-N,N-dimethylethanamine |
| Synonyms | trans-Tamoxifen; Crisafeno; Diemon; Tamoxifene; NSC-180973, Citofen; Istubol; ICI 46474; Nolvadex; ICI-46474; ICI46474; NSC 180973; tamoxifen; tamoxifeni citras; Novaldex |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: This product requires protection from light (avoid light exposure) during transportation and storage. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | ER/Estrogen receptor; HSP90 |
| ln Vitro | Tamoxifen (ICI 47699) does not influence MDA-MB-231 cells, but it has a significant inhibitory effect on MCF-7 cells (EC50=1.41 μM) and a lessened inhibitory effect on T47D cells (EC50=2.5 μM) [2]. |
| ln Vivo |
Gene knockout occurs when premutation mice receive an injection of tamoxifen (75 mg/kg; administered every five days at 6 weeks of age) which causes floxed exon excision [3]. The Tamoxifen-inducible gene knockout strategy has clear advantages in that expression of a gene can be ablated in adult mice at will in a tissue specific manner. To study the role of Med1 in adult heart, 7-week old TmcsMed1-/- mice are given a daily Iintraperitoneal injection of Tamoxifen at a dose of 65 mg/kg for 5 days and killed at selected intervals thereafter. qPCR analysis of RNA shows that the Med1 expression begin to decrease after 3 days of Tamoxifen injection (about 70% decrease), and by 5 days of injection, Med1 expression is almost non-detectable in the heart. Tamoxifen-inducible cardiac-specific disruption of Med1 (TmcsMed1-/-) in adult mice causes dilated cardiomyopathy[8]. |
| Enzyme Assay | Tamoxifen is a selective estrogen receptor modulator widely used for the prophylactic treatment of breast cancer. In addition to the estrogen receptor (ER), tamoxifen binds with high affinity to the microsomal antiestrogen binding site (AEBS), which is involved in ER-independent effects of tamoxifen. In the present study, we investigate the modulation of the biosynthesis of cholesterol in tumor cell lines by AEBS ligands. As a consequence of the treatment with the antitumoral drugs tamoxifen or PBPE, a selective AEBS ligand, we show that tumor cells produced a significant concentration- and time-dependent accumulation of cholesterol precursors. Sterols have been purified by HPLC and gas chromatography, and their chemical structures determined by mass spectrometric analysis. The major metabolites identified were 5alpha-cholest-8-en-3beta-ol for tamoxifen treatment and 5alpha-cholest-8-en-3beta-ol and cholesta-5,7-dien-3beta-ol, for PBPE treatment, suggesting that these AEBS ligands affect at least two enzymatic steps: the 3beta-hydroxysterol-Delta8-Delta7-isomerase and the 3beta-hydroxysterol-Delta7-reductase. Steroidal antiestrogens such as ICI 182,780 and RU 58,668 did not affect these enzymatic steps, because they do not bind to the AEBS. Transient co-expression of human 3beta-hydroxysterol-Delta8-Delta7-isomerase and 3beta-hydroxysterol-Delta7-reductase and immunoprecipitation experiments showed that both enzymes were required to reconstitute the AEBS in mammalian cells. Altogether, these data provide strong evidence that the AEBS is a hetero-oligomeric complex including 3beta-hydroxysterol-Delta8-Delta7-isomerase and the 3beta-hydroxysterol-Delta7-reductase as subunits that are necessary and sufficient for tamoxifen binding in mammary cells. Furthermore, because selective AEBS ligands are antitumoral compounds, these data suggest a link between cholesterol metabolism at a post-lanosterol step and tumor growth control. These data afford both the identification of the AEBS and give new insight into a novel molecular mechanism of action for drugs of clinical value[5]. |
| Cell Assay | Previous studies have shown that a styrylpyrone derivative (SPD) from a local tropical plant had antiprogestin and antiestrogenic effects in early pregnant mice models (Azimahtol et al. 1991). Antiprogestins and antiestrogens can be exploited as a therapeutic approach to breast cancer treatment and thus the antitumor activity of SPD was tested in three different human breast cancer cell lines that is: MCF- 7, T47D and MDA-MB-231, employing, the antiproliferative assay of Lin and Hwang (1991) slightly modified. SPD (10(-10) - 10(-6) M) exhibited strong antiproliferative activity in estrogen and progestin-dependent MCF-7 cells (EC50 = 2.24 x 10(-7) M) and in hormone insensitive MDA-MB-231 (EC50 = 5.62 x 10(-7) M), but caused only partial inhibition of the estrogen- insensitive T47D cells (EC50 = 1.58 x 10(-6) M). However, tamoxifen showed strong inhibition of MCF-7 cells (EC50 = 1.41 x 10(-6) M) and to a lesser extent the T47D cells (EC50 = 2.5 x 10(-6) M) but did not affect the MDA-MB-231 cells. SPD at 1 microM exerted a beffer antiestrogenic activity than 1 microM tamoxifen in suppressing the growth of MCF-7 cells stimulated by 1 nM estradiol. Combined treatment of both SPD and tamoxifen at 1 microM showed additional inhibition on the growth of MCF-7 cells in culture. The antiproliferative properties of SPD are effective on both receptor positive and receptor negative mammary cancer cells, and thus appear to be neither dependent on cellular receptor status nor cellular hormone responses. This enhances in vivo approaches as tumors are heterogenous masses with varying receptor status[2]. |
| Animal Protocol |
Animal/Disease Models: Aldh1l1-cre/ERT2 x Ai95 mice[3] Doses: 75 mg/kg Route of Administration: Injected for 5 days at 6 weeks of age Experimental Results: Resulted in the excision of the floxed exon and a gene knockout. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion An oral dose of 20mg reaches a Cmax of 40ng/mL with a Tmax of 5 hours. The metabolite N-desmethyltamoxifen reaches a Cmax of 15ng/mL. 10mg of tamoxifen orally twice daily for 3 months results in a Css of 120ng/mL and a Css of 336ng/mL. Tamoxifen is mainly eliminated in the feces. Animal studies have shown 75% of radiolabelled tamoxifen recovered in the feces, with negligible collection from urine. However, 1 human study showed 26.7% recovery in the urine and 24.7% in the feces. The volume of distribution of tamoxifen is approximately 50-60L/kg. The clearance of tamoxifen was 189mL/min in a study of six postmenopausal women. Tamoxifen appears to be absorbed slowly following oral administration, with peak serum concentrations generally occurring about 3-6 hours after a single dose. The extent of absorption in humans has not been adequately determined, but limited data from animal studies suggest that the drug is well absorbed. Data from animal studies also suggest that tamoxifen and/or its metabolites undergo extensive enterohepatic circulation. Following oral administration, peak serum tamoxifen concentrations average about 17 ng/mL after a single 10-mg dose, about 40 ng/mL after a single 20-mg dose, and 65-70 ng/mL after a single 40-mg dose; however, there is considerable interindividual variation in serum tamoxifen concentrations attained after single doses and at steady state with continuous dosing. Following a single oral dose of tamoxifen, peak serum concentrations of N-desmethyltamoxifen, the major metabolite of the drug, generally range from about 15-50% those of unchanged tamoxifen; however, with continuous dosing, steady-state serum concentrations of N-desmethyltamoxifen generally range from about 1-2 times those of unchanged tamoxifen. Following continuous administration in patients receiving oral tamoxifen 10 mg twice daily for 3 months, steady-state plasma concentrations of tamoxifen and N-desmethyltamoxifen average about 120 ng/mL (range: 67-183 ng/mL) and 336 ng/mL (range: 148-654 ng/mL), respectively. Steady-state serum concentrations of tamoxifen are generally attained after 3-4 weeks of continuous dosing, while those of N-desmethyltamoxifen are generally attained after 3-8 weeks of continuous dosing. Steady-state serum concentrations can be attained more rapidly with a loading-dose regimen, but there is no therapeutic advantage with such a regimen. For more Absorption, Distribution and Excretion (Complete) data for TAMOXIFEN (9 total), please visit the HSDB record page. Metabolism / Metabolites Tamoxifen can by hydroxylated to α-hydroxytamoxifen which is then glucuronidated or undergoes sulfate conjugation by sulfotransferase 2A1. Tamoxifen can also undergo N-oxidation by flavin monooxygenases 1 and 3 to tamoxifen N-oxide. Tamoxifen is N-dealkylated to N-desmethyltamoxifen by CYP2D6, CYP1A1, CYP1A2, CYP3A4, CYP1B1, CYP2C9, CYP2C19, and CYP3A5. N-desmethyltamoxifen can be sulfate conjugated to form N-desmethyltamoxifen sulfate, 4-hydroxylated by CYP2D6 to form endoxifen, or N-dealkylated again by CYP3A4 and CYP3A5 to N,N-didesmethyltamoxifen. N,N-didesmethyltamoxifen undergoes a substitution reaction to form tamoxifen metabolite Y, followed by ether cleavage to metabolite E, which can then be sulfate conjugated by sulfotransferase 1A1 and 1E1 or O-glucuronidated. Tamoxifen can also by 4-hydroxylated by CYP2D6, CYP2B6, CYP3A4, CYP2C9, and CYP2C19 to form 4-hydroxytamoxifen. 4-hydroxytamoxifen can undergo glucuronidation by UGT1A8, UGT1A10, UGT2B7, and UGT2B17 to tamoxifen glucuronides, sulfate conjugation by sulfotransferase 1A1 and 1E1 to 4-hydroxytamoxifen sulfate, or N-dealkylation by CYP3A4 and CYP3A5 to endoxifen. Endoxifen undergoes demethylation to norendoxifen, a reversible sulfate conjugation reaction via sulfotransferase 1A1 and 1E1 to 4-hydroxytamoxifen sulfate, sulfate conjugation via sulfotransferase 2A1 to 4-endoxifen sulfate, or glucuronidation via UGT1A8, UGT1A10, UGT2B7, or UGT2B15 to tamoxifen glucuronides. Tamoxifen is extensively metabolized after oral administration. N-desmethyl tamoxifen is the major metabolite found in plasma. N-desmethyl tamoxifen activity is similar to tamoxifen. 4-Hydroxytamoxifen and a side chain primary alcohol derivative of tamoxifen have been identified as minor metabolites in plasma. Tamoxifen is a substrate of cytochrome P450 CYP3A, CYP2C9 and CYP2D6, and an inhibitor of P-glycoprotein. Tamoxifen is rapidly and extensively metabolized, principally by demethylation and to a small degree by subsequent deamination and also by hydroxylation. Initial studies suggested that 4-hydroxytamoxifen (metabolite B) was the major metabolite of the drug, but subsequent studies using improved assay methodologies have shown that 4-hydroxytamoxifen is a minor metabolite and that the major metabolite is N-desmethyltamoxifen (metabolite X). The biologic activity of N-desmethyltamoxifen appears to be similar to that of tamoxifen. N-Desmethyltamoxifen undergoes demethylation to form N,N-desdimethyltamoxifen (metabolite Z) which undergoes subsequent deamination to form the primary alcohol metabolite (metabolite Y). Both 4-hydroxytamoxifen and a side chain primary alcohol derivative of tamoxifen have been identified as minor metabolites in plasma. 3,4-Dihydroxytamoxifen and an unidentified metabolite (metabolite E) also have been detected in plasma in small amounts. With continuous administration of tamoxifen, serum concentrations of N-desmethyltamoxifen are generally about 1-2 times those of unchanged tamoxifen, while those of N,N-desdimethyltamoxifen are about 20-40% those of unchanged tamoxifen and those of the primary alcohol metabolite are about 5-25% those of unchanged tamoxifen; concentrations of the hydroxylated metabolites and metabolite E appear to be less than 5% of those of unchanged tamoxifen. Several metabolites of tamoxifen, including 4-hydroxy-N-desmethyltamoxifen, 4-hydroxytamoxifen, N-desmethyltamoxifen, the primary alcohol, and N-desdimethyltamoxifen were identified and their concn determined in fluids and feces from patients receiving chronic tamoxifen treatment. The biological samples investigated were serum, pleural, pericardial and peritoneal effusions, cerebrospinal fluid, saliva, bile, feces, and urine. In serum, tamoxifen itself, and the metabolites N-desmethyltamoxifen and N-desdimethyltamoxifen were the prevailing species, but significant amounts of the metabolites the primary alcohol, 4-hydroxytamoxifen, 4-hydroxy-N-desmethyltamoxifen were also detected. About 3 hr after drug intake tamoxifen as well as, N-desmethyltamoxifen, an N-desdimethyltamoxifen) showed a peak in serum. This may be explained by efficient metabolism of the metabolite precursor before being distributed to peripheral compartments. Upon drug withdrawal all metabolites showed first-order elimination curves which paralleled that of tamoxifen suggesting that their rate of elimination exceeded that of tamoxifen and that the serum levels are production rate limited. The protein binding of tamoxifen and its major serum metabolites (the primary alcohol, N-desmethyltamoxifen, N-desdimethyltamoxifen) was determined and found to be higher than 98%. Albumin was the predominant carrier for tamoxifen in human plasma. The concn of tamoxifen and its metabolites in pleural, pericardial, and peritoneal effusions equalled those detected in serum, corresponding to an effusion/serum ratio between 0.2 and 1. Only trace amounts of tamoxifen and metabolite N-desmethyltamoxifen were detected in cerebrospinal fluid (CSF/serum ratio less than 0.02). In saliva, concn of tamoxifen and N-desmethyltamoxifen exceeded the amounts of free drug in serum, suggesting active transport or trapping of these compounds in the salivary gland. Bile and urine were rich in the hydroxylated, conjugated metabolites (the primary alcohol, 4-hydroxytamoxifen, 4-hydroxy-N-desmethyltamoxifen, whereas in feces unconjugated metabolite B and tamoxifen were the predominating species. The amount of tamoxifen, N-desmethyltamoxifen (metabolite X), N-desdimethyltamoxifen (metabolite Z), and hydroxylated metabolites (trans-1(4-beta-hydroxyethoxyphenyl)-1,2-diphenylbut-1-ene, 4-hydroxytamoxifen and 4-hydroxy-N-desmethyltamoxifen) were determined in brain metastases from breast cancer patients and in the surrounding brain tissues. Specimens were collected from the breast cancer patients who received tamoxifen for 7-180 days and with the last dose taken within 28 hr before surgical removal of the tumour. The concn of tamoxifen and its metabolites were up to 46 fold higher in the brain metastatic tumour and brain tissue than in serum. Metabolite N-desmethyltamoxifen was the most abundant species followed by tamoxifen and metabolite N-desdimethyltamoxifen. Small but significant amounts of the hydroxylated metabolites, trans-1(4-beta-hydroxyethoxyphenyl)-1,2-diphenylbut-1-ene, 4-hydroxytamoxifen and 4-hydroxy-N-desmethyltamoxifen were detected in most specimens. The ratios between the concn of tamoxifen and various metabolites were similar in tumour, brain and serum. This is the first report on the distribution of tamoxifen and metabolites into human brain and brain tumour, and the data form a basis for further investigation into the therapeutic effects of tamoxifen on brain metastases from breast cancer. Tamoxifen has known human metabolites that include 4'-Hydroxytamoxifen, alpha-Hydroxytamoxifen, 3-Hydroxytamoxifen, N-Desmethyltamoxifen, 4-Hydroxytamoxifen, and Tamoxifen N-glucuronide. Hepatic. Tamoxifen is extensively metabolized after oral administration. N-Desmethyl-tamoxifen is the major metabolite found in plasma. N-Desmethyl-tamoxifen's activity is similar to tamoxifen. 4-hydroxy-tamoxifen and a side chain primary alcohol derivative of tamoxifen have been identified as minor metabolites in plasma. 4-Hydroxy-tamoxifen formation is catalyzed mainly by cytochrome P450 (CYP) 2D6, and also by CYP2C9 and 3A4. At high tamoxifen concentrations, CYP2B6 also catalyzes 4-hydroxylation of the parent drug. 4-Hydroxy-tamoxifen possesses 30- to 100-times greater affinity for the estrogen receptor and 30- to 100-times greater potency at inhibiting estrogen-dependent cell proliferation compared to tamoxifen. It is also metabolized by flavin monooxygenases FMO1 and FMO3 to form tamoxifen-N-oxide. Route of Elimination: 65% of the dose was excreted from the body over 2 weeks in which fecal excretion was the primary route of elimination. Tamoxifen is excreted mainly as polar conjugates, with unchanged drug and unconjugated metabolites accounting for less than 30% of the total fecal radioactivity. Half Life: The decline in tamoxifen plasma concentrations is biphasic with a terminal elimination half-life of approximately 5 to 7 days. The estimated half-life of N-desmethyl tamoxifen is 14 days. Biological Half-Life The terminal elimination half-life of tamoxifen is 5 to 7 days, while the half-life of N-desmethyltamoxifen, the primary circulating metabolite, is approximately 14 days. Limited data suggest that tamoxifen has a distribution half-life of 7-14 hours and an elimination half-life of about 5-7 days (range: 3-21 days). The elimination half-life of N-desmethyltamoxifen, the major metabolite, is estimated to be 9-14 days. |
| Toxicity/Toxicokinetics |
Toxicity Summary IDENTIFICATION: Tamoxifen is an anti-estrogenic non-steroidal drug. Indications: Treatment of advanced breast cancer and adjuvant treatment of early breast cancer. Treatment of anovulatory infertility. HUMAN EXPOSURE: Main risks and target organs: Adverse effects in therapeutic use are usually mild. They include effects caused by antagonism of endogenous oestrogens: hot flushes, non-specific gastrointestinal effects (nausea and vomiting), central nervous system effects, and rare ocular effects. Adverse hematological effects have been reported, also isolated cases of death from peliosis hepatis and from hyperlipidemia. In the treatment of breast cancer, hypercalcemia and tumor flare can occur. Summary of clinical effects: Anti-estrogenic effects in women treated with tamoxifen include vasomotor symptoms (hot flushes), vaginal bleeding and (in premenopausal women) irregular menses, and pruritus vulvae. Nausea and vomiting can occur. Dizziness, lethargy, depression, irritability and cerebellar dysfunction have been described. Reversible retinopathy with macular edema has been reported after high cumulative doses, and corneal changes can occur. Thrombocytopenia or leukopenia have been associated with tamoxifen treatment. Thromboembolism, which may be due to the disease rather than the treatment, has been recorded in women given tamoxifen for breast cancer. Contraindications: Pregnancy is an absolute contraindication because of the anti-estrogenic effects. Routes of entry: Oral: Usual route of entry Absorption by route of exposure: Peak concentrations occur 4-7 hr after oral dosing. Peak concentrations after single oral dose are about 40 u/l. Distribution by route of exposure: Tamoxifen is more than 99% protein-bound in serum, predominantly to albumin. In patients with breast cancer, concentrations of tamoxifen and its metabolites in pleural, pericardial and peritoneal effusion fluid are between 20 and 100% of those in serum, but only trace amounts enter the cerebrospinal fluid. Concentrations in breast cancer tissue exceed those in serum. The volume of distribution is 50-60 l/kg. Biological half-life by route of exposure: The elimination is biphasic, with an initial half-life of around 7 hr and a terminal half-life of 7-11 days. Metabolism: Tamoxifen citrate undergoes extensive hepatic metabolism to: 1-(4-ethanolyloxyphenyl)-1,2-diphenylbut-1-ene (the primary alcohol), N-desmethyl tamoxifen, 4-hydroxy tamoxifen, 4-hydroxy-N-desmethyl tamoxifenn and N-desdimethyl tamoxifen Elimination by route of exposure: The major excretory route is via the bile as metabolites and enterohepatic recirculation occurs. Less than 1% is excreted in the urine. Mode of action: Toxicodynamics: The adverse effects observed are due mainly to its anti-estrogen effect, as Tamoxifen and certain of its metabolites antagonise the effects of estrogens in estrogen sensitive tissues. Pharmacodynamics: Tamoxifen and several of its metabolites (particularly 4-hydroxytamoxifen) bind to nuclear estrogen receptors in estrogen sensitive tissues, and also to a microsomal protein termed the anti-estrogen binding site. Tamoxifen interferes with the physiological sequence by which estrogen binds to its receptor, is translocated in the nucleus and then activates messenger RNA synthesis. Although the tamoxifen receptor complex is transported in the nucleus in the same way as estrogen receptor complex, it fails to activate synthesis of mRNA. Carcinogenicity: A case-control study showed a significantly increased relative risk of carcinoma of the uterus in women previously treated with tamoxifen and who had previously had radiotherapy involving the uterus. The study showed an increase in relative risk with tamoxifen treatment alone which was not statistically significant. Teratogenicity: Studies in neonatal male and female mice at relative doses 10 times higher than those used in humans have shown genital tract abnormalities. Interactions: Tamoxifen potentiates the anticoagulant effect of warfarin, and this interaction can be life-threatening. Main adverse effects: Adverse effects are usually mild. Thrombocytopenia, leukopenia, thromboembolism, peliosis hepatis and hyperlipidaemia have been mentioned in case reports. Severe hypercalcemia can occur rarely when treatment is started in patients with metastases to bone. Chronic poisoning: Ingestion: Retinal damage and keratitis have been reported in patients after large cumulative doses of tamoxifen, for more than 1 year, though sometimes with smaller doses. There seems to be correlation between long term tamoxifen administration and endometrial proliferation. Neurological: CNS: A case of depression, syncope, and incoordination has been described during therapy with 10 mg twice daily. The symptoms resolved when tamoxifen was discontinued and reappeared when treatment was restarted. Gastrointestinal: Nausea and vomiting occur with therapeutic doses in some patients, and are anticipated in overdosage. Hepatic: A fatal case of peliosis hepatis has been reported in a woman treated with tamoxifen for 2 years after mastectomy for carcinoma. Urinary: Other: A case of persistent nocturnal priapism has been reported. Endocrine and reproductive systems: The anti-estrogenic effects of tamoxifen in premenopausal women receiving therapeutic doses can cause irregular menses. Anti-estrogenic adverse effects in women treated with tamoxifen include vasomotor symptoms and vaginal bleeding and pruritus vulvae. Eye, ear, nose, throat: local effects: Treatment has been associated with retinal and corneal changes. Hematological: Thromboembolism may be more common in patients treated with tamoxifen, though this is not certain, as patients with cancer are at increased risk anyway. A small reduction in antithrombin III concentration was noted in a study of 11 postmenopausal women treated with tamoxifen, but it was clinically insignificant, and no significant reduction was seen in a group of premenopausal women. Thrombocytopenia and leukopenia can occur during therapy, but are not usually severe. One case of severe myelosuppression has been reported. Fluid and electrolyte disturbances: Severe hypercalcemia, associated with increased bone resorption, has been noted when patients with bony metastases commenced therapy. Others: Severe hyperlipidemia is occasionally seen, and has been ascribed to an estrogenic effect. Special risks: Pregnancy, breast feeding and enzyme deficiencies. ANIMAL/PLANT STUDIES: In some animal species, estrogenic agonist effects become manifest at dosages equivalent to 10-100 times the human therapeutic dose. Mutagenicity: Tamoxifen is believed not to be mutagenic. /Tamoxifen citrate/ Tamoxifen is a nonsteroidal agent that binds to estrogen receptors (ER), inducing a conformational change in the receptor. This results in a blockage or change in the expression of estrogen dependent genes. The prolonged binding of tamoxifen to the nuclear chromatin of these results in reduced DNA polymerase activity, impaired thymidine utilization, blockade of estradiol uptake, and decreased estrogen response. It is likely that tamoxifen interacts with other coactivators or corepressors in the tissue and binds with different estrogen receptors, ER-alpha or ER-beta, producing both estrogenic and antiestrogenic effects. Interactions Concomitant use of Aminoglutethimide may decrease plasma concentrations of tamoxifen and N-desmethyl tamoxifen. Concomitant use coumarin-derivatived anticoagulants, may result in significant increase in anticoagulant effect; use is contraindicated in women for reducing the risk of breast cancer in high-risk women and women with ductal carcinoma in situ (DCIS). Concomitant use of bromocriptine may increase serum levels of tamoxifen and N-desmethyl tamoxifen. Concomitant use of cytotoxic agents may result in increased risk of thromboembolic events. For more Interactions (Complete) data for TAMOXIFEN (12 total), please visit the HSDB record page. |
| References |
[1]. Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med. 1998 Nov 26;339(22):1609-18. [2]. Hawariah A, et al. In vitro response of human breast cancer cell lines to the growth-inhibitory effects of styrylpyrone derivative (SPD) and assessment of its antiestrogenicity. Anticancer Res. 1998 Nov-Dec;18(6A):4383-6. [3]. Jun Nagai, et al. Hyperactivity with Disrupted Attention by Activation of an Astrocyte Synaptogenic Cue. Cell. 2019 May 16;177(5):1280-1292.e20. [4]. Zhao R, et al. Tamoxifen enhances the Hsp90 molecular chaperone ATPase activity. PLoS One. 2010 Apr 1;5(4):e9934. [5]. Kedjouar B, et al. Molecular characterization of the microsomal tamoxifen binding site. J Biol Chem. 2004 Aug 6;279(32):34048-61. [6]. Feil S, et, al. Inducible Cre mice. Methods Mol Biol. 2009;530:343-63. [7]. Laura Cooper, et al. Screening and Reverse-Engineering of Estrogen Receptor Ligands as Potent Pan-Filovirus Inhibitors. J Med Chem. 2020 Sep 4. [8]. Cardiomyocyte-Specific Ablation of Med1 Subunit of the Mediator Complex Causes Lethal DilatedCardiomyopathy in Mice. PLoS One. 2016 Aug 22;11(8):e0160755. |
| Additional Infomation |
Therapeutic Uses Anticarcinogenic Agents; Antineoplastic Agents, Hormonal; Carcinogens; Estrogen Antagonists Antiestrogen; antineoplastic (hormonal). Tamoxifen is indicated for adjuvant treatment of axillary nodenegative breast cancer in women following total mastectomy or segmental mastectomy, axillary dissection, and breast irradiation. Data are insufficient to predict which women are most likely to benefit and to determine if tamoxifen provides any benefit in women with tumors of less than 1 cm. Tamoxifen is /also/ indicated for adjuvant treatment of axillary node-positive breast cancer in postmenopausal women following total mastectomy or segmental mastectomy, axillary dissection, and breast irradiation. In some tamoxifen adjuvant studies, most of the benefit to date has been in the subgroup with four or more positive axillary nodes. /Include in US product label/ Tamoxifen is indicated to reduce the risk of developing breast cancer in women who have been determined to be at high risk for developing this cancer. A woman is considered to be at high risk if she is at least 35 years of age and has a 5-year predicted risk of developing breast cancer greater than or equal to 1.67%. /Included in US product label/ For more Therapeutic Uses (Complete) data for TAMOXIFEN (8 total), please visit the HSDB record page. Drug Warnings Cases of tamoxifen-associated hepatotoxicity have been described, including cholestasis with or without cytolysis and steatohepatitis. We report the case of a female patient who developed hepatic alterations while undergoing continuous tamoxifen treatment. There are recent reports of postmenopausal bleeding from endometrial polyps in women receiving tamoxifen therapy for breast cancer. /The authors/ describe four additional patients who presented with vaginal bleeding, and emphasize the pathology. These polyps demonstrated cystically dilated glands in all cases and stromal decidualization in two; in one instance, metastatic breast carcinoma was present in the polyp. The mechanisms by which tamoxifen may affect the development of these polyps are discussed. This case report serves to emphasize two important features of metastatic breast carcinoma. First, that tamoxifen-induced flare, although a rare and self-limiting phenomenon, may be fatal and must thus be recognized and treated promptly. Secondly, those patients presenting with hypercalcaemia, as part of tamoxifen-induced tumour flare, invariably have metastatic disease but they may enjoy a durable prognosis if this is confined to the skeleton. The fourth case of heterologous mesodermal tumour of the uterine corpus, that developed, years following tamoxifen therapy for breast cancer in a postmenopausal woman with no previous pelvic irradiation, is presented with coincidental endometriosis and endometrial intraepithelial carcinoma. For more Drug Warnings (Complete) data for TAMOXIFEN (42 total), please visit the HSDB record page. Pharmacodynamics Tamoxifen is a selective estrogen receptor modulator that inhibits growth and promotes apoptosis in estrogen receptor positive tumors. It has a long duration of action as the active metabolite N-desmethyltamoxifen has a half life of approximately 2 weeks. It has a narrow therapeutic index as higher doses can lead to breathing difficulty or convulsions. Tamoxifen administration is also associated with an increased incidence of uterine malignancies. |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: 5 mg/mL (13.46 mM) in 30% PEG400 0.5% Tween80 + 5% Propanediol 64.5%H2O (add these co-solvents sequentially from left to right, and one by one), suspension solution; with sonication. Solubility in Formulation 2: 2.5 mg/mL (6.73 mM) in 10% EtOH + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear EtOH stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (6.73 mM) (saturation unknown) in 10% EtOH + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear EtOH stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: ≥ 2.08 mg/mL (5.60 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL of PEG300 and mix evenly; then add 50 μL of Tween-80 to the above solution and mix evenly; then add 450 μL of normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 5: 2.08 mg/mL (5.60 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 6: ≥ 2.08 mg/mL (5.60 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 7: ≥ 0.83 mg/mL (2.23 mM) (saturation unknown) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 8: 0.83 mg/mL (2.23 mM) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 9: (saturation unknown) in Corn oil:40 mg/mL or 107.67 mM (add these co-solvents sequentially from left to right, and one by one), Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 10: 40 mg/mL (107.67 mM) in Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6917 mL | 13.4586 mL | 26.9172 mL | |
| 5 mM | 0.5383 mL | 2.6917 mL | 5.3834 mL | |
| 10 mM | 0.2692 mL | 1.3459 mL | 2.6917 mL |