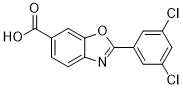
Tafamidis (also known as Fx-1006 or PF-06291826; Vyndaqel and Vyndamax), a potent and selective transthyretin (TTR) stabilizer, is a medication approved in several countries for use in delaying disease progression in adults with certain forms of transthyretin amyloidosis.
Physicochemical Properties
| Molecular Formula | C14H7CL2NO3 |
| Molecular Weight | 308.1163 |
| Exact Mass | 306.98 |
| Elemental Analysis | C, 54.58; H, 2.29; Cl, 23.01; N, 4.55; O, 15.58 |
| CAS # | 594839-88-0 |
| Related CAS # | Tafamidis meglumine;951395-08-7;Tafamidis-d3 |
| PubChem CID | 11001318 |
| Appearance | Typically exists as white to off-white solids at room temperature |
| Density | 1.5±0.1 g/cm3 |
| Boiling Point | 486.7±40.0 °C at 760 mmHg |
| Flash Point | 248.1±27.3 °C |
| Vapour Pressure | 0.0±1.3 mmHg at 25°C |
| Index of Refraction | 1.677 |
| LogP | 5.29 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 4 |
| Rotatable Bond Count | 2 |
| Heavy Atom Count | 20 |
| Complexity | 371 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | ClC1C([H])=C(C([H])=C(C=1[H])C1=NC2C([H])=C([H])C(C(=O)O[H])=C([H])C=2O1)Cl |
| InChi Key | TXEIIPDJKFWEEC-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C14H7Cl2NO3/c15-9-3-8(4-10(16)6-9)13-17-11-2-1-7(14(18)19)5-12(11)20-13/h1-6H,(H,18,19) |
| Chemical Name | 2-(3,5-dichlorophenyl)benzo[d]oxazole-6-carboxylic acid |
| Synonyms | Fx-1006, PF06291826; Fx1006, PF-06291826; Fx 1006, Fx-1006A; PF 06291826; Tafamidis; Vyndaqel; TAFAMIDIS; 594839-88-0; Vyndamax; FX-1006; 2-(3,5-Dichlorophenyl)-1,3-Benzoxazole-6-Carboxylic Acid; 2-(3,5-Dichlorophenyl)-6-benzoxazole carboxylic acid; tafamidisum; 8FG9H9D31J; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
TTR (transthyretin) (EC50 = 2.7-3.2 μM) Transthyretin (TTR) tetramer (binds with negative cooperativity to the two thyroxine-binding sites; \(K_{d1} \sim 2-3 \text{ nM}\), \(K_{d2} \sim 154-278 \text{ nM}\); EC\(_{50}\) ~2.7–3.2 µM for fibril inhibition under acidic conditions) [1] |
| ln Vitro |
TTR is kinetically stabilized when tacamimeis binds to the two tetramer's typically conserved polarin binding sites with negative coupling (Kd = ∼2 nM and ∼200 nM) [1]. After 72 hours at =4.4-4.5, tacramids (0-7.2 μM) dose-dependently suppresses WT-TTR amyloidosis [1]. Tafamidis dose-dependently inhibits amyloid fibril formation by wild-type (WT), V30M, and V122I TTR homotetramers under acidic, fibril-promoting conditions (pH 4.4–4.5, 72 h incubation), with EC\(_{50}\) values corresponding to a tafamidis:TTR molar ratio of <1. Tafamidis kinetically stabilizes TTR tetramers against urea-mediated denaturation (6.5 M urea, 72 h), reducing tetramer dissociation to less than 3% at a 2:1 molar ratio. Tafamidis selectively binds to TTR in human plasma with a stoichiometry of ~0.81 ± 0.02 tafamidis per TTR tetramer, indicating high selectivity among plasma proteins. Tafamidis stabilizes a broad range of pathogenic TTR variants (V30M, Y69H, F64S, I84S, V122I, L111M) in plasma under urea denaturation stress.[1] |
| ln Vivo |
ATTR amyloidosis is a systemic, debilitating and fatal disease caused by transthyretin (TTR) amyloid accumulation. RNA interference (RNAi) is a clinically validated technology that may be a promising approach to the treatment of ATTR amyloidosis. The vast majority of TTR, the soluble precursor of TTR amyloid, is expressed and synthesized in the liver. RNAi technology enables robust hepatic gene silencing, the goal of which would be to reduce systemic levels of TTR and mitigate many of the clinical manifestations of ATTR that arise from hepatic TTR expression. To test this hypothesis, TTR-targeting siRNAs were evaluated in a murine model of hereditary ATTR amyloidosis. RNAi-mediated silencing of hepatic TTR expression inhibited TTR deposition and facilitated regression of existing TTR deposits in pathologically relevant tissues. Further, the extent of deposit regression correlated with the level of RNAi-mediated knockdown. In comparison to the TTR stabilizer, tafamidis, RNAi-mediated TTR knockdown led to greater regression of TTR deposits across a broader range of affected tissues. Together, the data presented herein support the therapeutic hypothesis behind TTR lowering and highlight the potential of RNAi in the treatment of patients afflicted with ATTR amyloidosis[2]. In a phase II/III clinical trial of tafamidis in V30M TTR-FAP patients, this kinetic stabilizer demonstrated clinical efficacy over 18 mo of treatment. Relative to placebo controls, patients receiving tafamidis had 52% less neurologic deterioration, 53% and 80% preservation of large- and small-nerve fiber function, and improved nutritional status, outcomes that are associated with an improved quality of life. The tafamidis preclinical data presented within, when considered in concert with the clinical efficacy data, provide unique pharmacologic evidence supporting the amyloid hypothesis, the notion that lowering the efficiency of the amyloid cascade halts the degeneration of the peripheral and autonomic nervous system[1]. In a Phase II/III clinical trial in V30M TTR-FAP patients, tafamidis treatment over 18 months resulted in 52% less neurological deterioration, 53–80% preservation of nerve fiber function, and improved nutritional status compared to placebo.[1] |
| Enzyme Assay |
Tafamidis Binds with High Affinity to TTR at Its T4-Binding Sites. Tafamidis Stabilizes the Weaker TTR Dimer–Dimer Interface. Tafamidis Binds Selectively to TTR in Human Plasma. Tafamidis Stabilizes WT, V30M, and V122I TTR in Human Plasma. Tafamidis Stabilizes a Broad Range of Pathogenic TTR Variants. Immunoturbidity Assay for Stabilization of TTR Tetramer in Human Plasma.[1] Urea denaturation of TTR in human plasma and chemical crosslinking was performed as described (see text and Fig. 6.) with minor modifications, except that TTR was quantified by immunoturbidity. Human plasma samples were thawed on ice and insoluble material was removed by centrifugation. For each, 4 µL was removed, and the initial TTR concentrations were determined by immunoturbidity. For each stabilization determination, 80 µL aliquots of each plasma sample were retained and 1.6 µL of either 5% dimethyl sulfoxide (DMSO) or 360 µM tafamidis in 5% DMSO was added. After incubation at room temperature for 15 minutes, 120 µL of urea buffer (8 M urea, 40 mM sodium phosphate, 80 mM KCl, pH 7.4) was added and samples were mixed and incubated at room temperature for the indicated time (typically 48 h). All samples were cross-linked with 3.2 µL of 25% glutaraldehyde. After 4 minutes, the reaction was quenched with 5.6 µL of 1.85 M NaBH4 (freshly prepared in 0.1 N NaOH) and incubated for 5 minutes. Postdenaturation TTR concentrations (4 µL) were determined by immunoturbidity. Olympus OSR6175 reagent and Prealbumin Calibrator ODR3029 were used according to the manufacturers’ instructions. To assess the correlation between the two detection methods, we analyzed plasma samples after urea treatment and glutaraldehyde crosslinking in parallel by Western blot and immunoturbidity. In the control samples, the amount of TTR detected by immunoturbidity decreased from an initial value of 22 mg/dL to 3 mg/dL after 3 d in urea. In the presence of tafamidis, 13 mg/dL of TTR remained; a level that was in good agreement with results from the Western blot assay (Fig. S3A). Isothermal titration calorimetry (ITC) was used to determine binding constants. A solution of tafamidis was titrated into a cell containing WT-TTR (17 µM) at 25°C. Heat changes were measured and integrated to generate a binding isotherm, which was fit to a negative cooperativity model yielding \(K_{d1} = 3 \text{ nM}\) and \(K_{d2} = 278 \text{ nM}\).[1] Subunit exchange experiments at physiologic pH were used to indirectly determine tetramer dissociation rates and calculate binding constants (\(K_{d1} = 2 \text{ nM}\), \(K_{d2} = 154 \text{ nM}\)) by analyzing the fraction of unbound TTR tetramers as a function of tafamidis concentration.[1] |
| Animal Protocol |
Evaluation of tafamidis in hTTR V30M HSF1± mice[2] Tafamidis/meglumine (tafamidis) and its respective meglumine only control (meglumine) were prepared as previously described. Four hundred microliters of 2 mg/ml tafamidis (0.8 mg total) or its respective meglumine control were administered via subcutaneous injection to 15-month-old hTTR V30M HSF1± mice on days 0, 3, 5, 7, 10, 12, 14, 17, 19, 21, 24, 26, 28, 31, 33, 35 and 38. TTR tissue deposition was evaluated on day 52 as described earlier. To confirm tafamidis-mediated stabilization of serum TTR, serum TTR tetramer stability was analyzed on days -7, 9, 23 and 37 using a modified version of a previously described TTR tetramer stability assay. See Supplementary Figure 2 for more detail on assay conditions and tetramer detection and quantitation. To quantify the extent of stabilization, % TTR tetramer stabilization was calculated using the following equation as previously described. To compare the efficacy of the tetramer stabilization approach to that of RNAi-mediated TTR knockdown, we evaluated tafamidis in the hTTR V30M HSF1± model and quantified the impact of TTR tetramer stabilization on the regression of preexisting TTR deposits. To compensate for differences in dose frequency and route of administration, mice were administered excess tafamidis (>100× on mg/kg basis) to enable sufficient TTR tetramer stabilization. Although administration of tafamidis resulted in a significant and clinically relevant degree of serum TTR tetramer stabilization, only moderate TTR deposit regression was observed in the sciatic nerve and dorsal root ganglion; consistent regression was not observed in other tissues examined. It should be noted that the study duration was chosen to allow a more direct comparison with siTTR1 (Figure 3) and, as such, it is possible that longer term administration of tafamidis may have resulted in greater deposit regression in the hTTR V30M HSF1± model. However, in these conditions, TTR lowering seems to be more effective. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Tafamidis reaches a Cmax of 1430.93ng/mL with a Tmax of 1.75h fasted and 4h fed. The AUC of tafamidis is 47,864.31ng\h/mL. A 20mg oral dose of tafamidis is approximately 59% recovered in the feces, largely as unchanged drug. Approximately 22% of a 20mg oral dose is recovered in the urine, mostly as the glucuronide metabolite. The apparent volume of distribution at steady state is 18.5L. The oral clearance of tafamidis is 0.263L/h. The apparent total clearance is 0.44L/h. Metabolism / Metabolites Tafamidis is largely not subject to first pass or oxidative metabolism, being 90% unchanged after in in vitro experiments. Preclinical data suggest tafamidis is mainly metabolized through glucuronidation and excreted in bile. Biological Half-Life The half life of tafamidis is 49h. Tafamidis is orally bioavailable and selectively binds TTR in blood without significant binding to other plasma proteins.[1] The plasma half-life of TTR is approximately 24 hours, and tafamidis stabilizes TTR over this period as shown in subunit exchange experiments.[1] |
| Toxicity/Toxicokinetics |
Protein Binding Tafamidis 99.9% protein bound in plasma, mostly to transthyretin. Tafamidis was well tolerated in clinical trials with no clinically relevant effects on thyroid function or laboratory measures, despite binding to thyroxine-binding sites on TTR.[1] |
| References |
[1]. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A, 2012. 109(24): p. 9629-34. |
| Additional Infomation |
Tafamidis is a member of the class of 1,3-benzoxazoles that is 1,3-benzoxazole-6-carboxylic acid in which the hydrogen at position 2 is replaced by a 3,5-dichlorophenyl group. Used (as its meglumine salt) for the amelioration of transthyretin-related hereditary amyloidosis. It has a role as a central nervous system drug. It is a member of 1,3-benzoxazoles, a monocarboxylic acid and a dichlorobenzene. It is a conjugate acid of a tafamidis(1-). Tafamidis and tafamidis meglumine (FX-1006A) are benzoxazole derivatives developed by FoldRX. Tafamidis is structurally similar to diflusinal. Tafamidis was granted an EMA market authorisation on 16 November 2011 and FDA approval on 3 May 2019. See also: Tafamidis Meglumine (has salt form). Drug Indication Tafamidis is indicated to treat cardiomyopathy of wild type or hereditary transthyretin-mediated amyloidosis in adults. FDA Label Mechanism of Action Genetic mutations or natural misfolding of transthyretin destabalizes transthyretin tetramers, leading to their dissociation and aggregation in tissues, and disrupting the normal function of these tissues. Tafamidis binds to transthyretin tetramers at the thyroxin binding sites, stabilizing the tetramer, reducing the availability of monomers for amyloidogenesis. Pharmacodynamics Tafamidis stabilizes transthyretin tetramers, reducing the amount of monomers available for amyloidogenesis. It has a long duration of action as it is given once daily, and a wide therapeutic window. Tafamidis is a benzoxazole derivative (2-(3,5-dichloro-phenyl)-benzoxazole-6-carboxylic acid) developed as an orally available TTR kinetic stabilizer to inhibit tetramer dissociation, the rate-limiting step in TTR amyloidogenesis. It mimics the protective effect of the T119M mutation, which slows tetramer dissociation and prevents disease in compound heterozygotes. The crystal structure of tafamidis bound to TTR (1.3 Å resolution) shows it occupies the thyroxine-binding sites, with hydrophobic interactions from dichloro-substituents and water-mediated H-bonds via its carboxylate group, stabilizing the dimer-dimer interface.[1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~37.5 mg/mL (~121.71 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: 2.5 mg/mL (8.11 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (8.11 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (8.11 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2455 mL | 16.2274 mL | 32.4549 mL | |
| 5 mM | 0.6491 mL | 3.2455 mL | 6.4910 mL | |
| 10 mM | 0.3245 mL | 1.6227 mL | 3.2455 mL |