TRi-1 (also know nas TXNRD1 inhibitor 1) is a novel, potent, specific and irreversible inhibitor of cytosolic thioredoxin reductase 1 (TXNRD1) with anticancer potential activity. It showed 5- to 10-fold higher specificity for TXNRD1 over TXNRD2.
Physicochemical Properties
| Molecular Formula | C12H9CLN2O5S |
| Molecular Weight | 328.728260755539 |
| Exact Mass | 327 |
| Elemental Analysis | C, 47.64; H, 3.08; Cl, 10.82; N, 4.27; O, 24.41; S, 9.78 |
| CAS # | 246020-68-8 |
| PubChem CID | 2801235 |
| Appearance | Light yellow to yellow solid powder |
| LogP | 2.7 |
| Hydrogen Bond Donor Count | 0 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 3 |
| Heavy Atom Count | 21 |
| Complexity | 467 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | PRKWNRUVAZZHPH-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C12H9ClN2O5S/c1-20-11-7-6-10(15(16)17)12(14-11)21(18,19)9-4-2-8(13)3-5-9/h2-7H,1H3 |
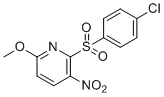
| Chemical Name | 2-(4-chlorophenyl)sulfonyl-6-methoxy-3-nitropyridine |
| Synonyms | TRi-1 (TXNRD1 inhibitor 1); TRi 1 (TXNRD1 inhibitor-1); TRi1 (TXNRD 1 inhibitor 1) |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: This product requires protection from light (avoid light exposure) during transportation and storage. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | In FaDu cells, TRi-1 (0.679 and 6.79 μM; 6 hours) potently stimulates JNK and p38 phosphorylation while having no effect on cellular glutathione (GSH) contents [1]. TRi-1 (2 μM) H2O2 is added to grown FaDu cells in a concentration- and time-dependent manner to create TRi-1 (0.1-10 μM; 0-10 h) [1]. TRi-1 at 10 nM. |
| ln Vitro |
In FaDu cells, TRi-1 (0.679 and 6.79 μM; 6 hours) potently stimulates JNK and p38 phosphorylation while having no effect on cellular glutathione (GSH) contents [1]. TRi-1 (2 μM) H2O2 is added to grown FaDu cells in a concentration- and time-dependent manner to create TRi-1 (0.1-10 μM; 0-10 h) [1]. TRi-1 at 10 nM. TRi-1 inhibits recombinant TXNRD1 enzyme activity irreversibly and in an NADPH-dependent manner. [1] Treatment with TRi-1 inhibits cellular TXNRD activity in cancer cell lines (e.g., FaDu) with equal or greater potency compared to auranofin. [1] TRi-1 treatment does not affect cellular glutathione (GSH) concentrations, whereas auranofin and TRi-2 lower GSH at high doses. [1] TRi-1 efficiently activates phosphorylation of JNK and p38 in FaDu cells, a downstream effect consistent with thioredoxin system inhibition. [1] TRi-1 converts inhibited TXNRD1 into pro-oxidant SecTRAPs (selenium compromised thioredoxin reductase-derived apoptotic proteins), which maintain NADPH-dependent redox cycling (e.g., with juglone) and lead to increased cellular H2O2 production. [1] TRi-1 has minimal effects on mitochondrial basal respiration and maximal respiratory capacity in HCT116 cells, unlike auranofin which severely impairs mitochondrial function. [1] TRi-1 exhibits cytotoxic activity against all cell lines in the NCI-60 panel, with an average growth inhibition to 50% (GI50) of 6.31 µM. Its cytotoxicity profile across the panel is least similar to that of auranofin. [1] TRi-1 shows preferential cytotoxicity towards cancer cells (e.g., A549 lung adenocarcinoma) over non-cancerous cells (e.g., mouse embryonic fibroblasts (MEFs), primary human fibroblasts, primary keratinocytes, CCD841 colon epithelial cells). MEFs with genetic deletion of Txnrd1 show additional resistance to TRi-1. [1] Cytotoxicity of TRi-1 is enhanced by co-treatment with buthionine sulphoximine (BSO), an inhibitor of GSH synthesis, supporting a mechanism involving sensitization to oxidative stress. [1] The cytotoxic potency of TRi-1 in a panel of human cancer cell lines (including HCT116 subclones with altered p53 or BCL-2 overexpression) shows minor influence from cancer cell genotype. [1] |
| ln Vivo |
TRi-1 (10 mg/kg; IV; twice daily for 4 days or 5 mg/kg; IP; twice weekly for 3 weeks) damages human tumor xenografts and syngeneic mice tumors In SCID mice bearing established human FaDu cell xenografts, intravenous (i.v.) administration of TRi-1 (10 mg/kg, twice daily for 4 days) significantly decreases tumor growth compared to vehicle controls within four days. [1] In the same FaDu xenograft model, treatment with TRi-1 (5 mg/kg, intraperitoneal (i.p.) once daily) leads to a marked decrease in [18F]-FDG uptake (measured by PET) in viable tumor regions after four days, while vehicle-treated tumors show increased uptake. Some tumors in the TRi-1-treated group develop low-uptake cores. [1] Excision and analysis of FaDu xenografts from mice treated with TRi-1 show increased staining for activated caspase-3 between days three and four of treatment. [1] In PyMT-MMTV transgenic mice that spontaneously develop malignant breast cancer, treatment with TRi-1 (5 mg/kg, i.p., twice a week for three weeks) significantly impairs tumor growth compared to vehicle controls. All mice in the TRi-1 group survive the observation period. [1] In athymic mice bearing orthotopic MDA-MB-231 human breast cancer xenografts, treatment with TRi-1 (10 mg/kg, administered either i.v. or i.p. on a schedule of daily for 5 days, then 3 times per week for 2 weeks) significantly decreases tumor growth compared to vehicle controls, with one tumor in the i.p. group completely regressing. [1] |
| Enzyme Assay |
Recombinant enzyme assays for TXNRD1 and GSR inhibition were performed in a reaction buffer containing 50 mM Tris pH 7.5 with 2 mM EDTA and 0.1 mg/mL bovine serum albumin. Assays were performed in triplicate in 96-well plates, monitoring activity via absorbance changes. Compounds were dissolved in DMSO. Recombinant rat TXNRD1 was used for initial screens, while human recombinant TXNRD1 and TXNRD2 were used for isoenzyme specificity comparisons. The TXNRD1 assay was adapted from a standard cuvette protocol using DTNB as a substrate or a TXN1-coupled insulin reduction assay. Activity was normalized to DMSO and no-enzyme controls. [1] To assess irreversible inhibition, TXNRD1 was incubated with compounds in the presence or absence of NADPH. The enzyme was then desalted to remove unbound compound and cofactors, and the remaining activity was measured. [1] SecTRAP activity was measured by pre-incubating NADPH-reduced TXNRD1 with inhibitors at concentrations that completely inhibit native activity. After desalting, the residual NADPH-dependent reduction of juglone was measured as an indicator of pro-oxidant gain-of-function. [1] Inhibition of glutathione peroxidase 1 (GPX1) was tested by preincubating the enzyme with compounds in the absence of GSH, followed by measurement of residual activity. [1] The specificity of inhibition in the context of cellular nucleophiles was tested by pre-incubating compounds with GSH before adding them to the TXNRD1 reaction mix, or by co-incubating compounds with TXNRD1, NADPH, and varying concentrations of GSH (and sometimes TXN1) before initiating the enzymatic reaction with insulin. [1] |
| Cell Assay |
Western Blot Analysis [1] Cell Types: FaDu Cell Tested Concentrations: 0.679 and 6.79 μM Incubation Duration: 6 h Experimental Results: Effective activation of JNK and p38 phosphorylation. -100 μM; 48 h) shows cytotoxicity to cancer cells [1]. Cytotoxicity assay[1] Cell Types: Leukemia, Non-Small Cell Lung Cancer, Central Nervous System, Colon, Melanoma, Ovarian, Kidney, Prostate, and Breast Cancer Cell Tested Concentrations: 10 nM-100 μM Incubation Duration: 48 hrs (hours) Experimental Results: Potency against each cell line tested was shown, with an average growth inhibition of 50% (GI50) of 6.31 μM. Cells were cultured at 37°C in 5% CO2 in medium containing penicillin/streptomycin, L-glutamine, 10% fetal bovine serum, and 25 nM sodium selenite. All experiments were performed in triplicate. Compounds were diluted in DMSO to a final concentration of 0.01%. [1] For cytotoxicity assays (cell viability), cells were incubated with compounds for 72 hours, and viability was determined. IC50/GI50 values were calculated from dose-response curves. [1] Cellular TXNRD activity was measured after treating cells (e.g., FaDu) with compounds for 3 hours. Cells were lysed, and enzyme activity was determined using an insulin-endpoint assay. [1] Cellular glutathione (GSH) concentrations were measured after exposing cells to compounds for 6 hours at a concentration of 10 times the IC50 for the respective cell line. [1] For analysis of signaling pathways, cells were treated with compounds for 6 hours, followed by lysis and Western blot analysis to detect phosphorylated and total levels of JNK and p38. [1] Cellular H2O2 production was measured using an Amplex red assay. Cells were treated with compounds, and H2O2 release into the medium was monitored fluorometrically in a concentration- and time-dependent manner. [1] Mitochondrial respiration was assessed using a Seahorse analyzer. Cells were seeded and treated with compounds for either 30 minutes or 5 hours. Oxygen consumption rate (OCR) was measured to determine parameters such as ATP-coupled basal respiration and maximal respiratory capacity. [1] Colony-forming assays were performed by treating cells with compounds, allowing them to form colonies over a period, then fixing, staining, and counting colonies to assess long-term clonogenic survival. [1] |
| Animal Protocol |
Animal/Disease Models: PyMT-MMTV mice that spontaneously develop malignant breast cancer tumors [1] Doses: 5 mg/kg Route of Administration: intraperitoneal (ip) injection, twice a week for 3 weeks Experimental Results: Impaired tumor growth, significant tumor volume Zoom out. A dose escalation toxicity study was performed in Fox Chase SCID mice. Mice were treated once with TRi-1 (0.7–10 mg/kg) via intravenous (i.v.) injection, and health status was observed for up to 72 hours. [1] For repeated dose toxicity and efficacy studies in tumor-bearing mice, SCID mice were inoculated subcutaneously with FaDu cells. After tumors were established (13 days), mice were treated i.v. twice daily for four days (total of nine injections over five days) with TRi-1 (10 mg/kg), TRi-2 (15 mg/kg), auranofin (10 mg/kg), or vehicle. Mouse health, weight, and tumor volume (by caliper) were monitored daily. [1] For PET imaging studies, SCID mice bearing FaDu xenografts underwent baseline [18F]-FDG PET imaging after 11 days of tumor growth. Subsequently, mice were treated once daily i.p. with TRi-1 (5 mg/kg) or vehicle. A follow-up PET scan was performed on day three of treatment. Mice were euthanized on day three or four for tumor excision and analysis (e.g., caspase-3 staining). [1] In the PyMT-MMTV spontaneous breast cancer model, transgenic mice were treated twice a week via i.p. injection with TRi-1 (5 mg/kg), auranofin (10 mg/kg), or vehicle for three weeks. Tumor volume in mammary glands was measured regularly by caliper. [1] In the MDA-MB-231 orthotopic xenograft model, athymic nude mice were inoculated with cells into the mammary fat pad. When tumors reached a specified volume, mice were randomized and treated with TRi-1 (10 mg/kg) via either i.v. or i.p. injection, or vehicle i.v., on a schedule of once daily for the first five days, then two days off, followed by three times per week for two weeks. Mouse health, weight, and tumor volume were monitored. [1] The compound TRi-1 was dissolved in DMSO and then formulated in a solution of 10% Cremophor EL, 10% ethanol, and 80% PBS for in vivo administration. [1] |
| Toxicity/Toxicokinetics |
TRi-1 was well tolerated in mice up to the highest administrable dose of 10 mg/kg i.v., with no overt signs of toxicity observed over a 72-hour period in a dose escalation study. [1] In repeated dose studies with tumor-bearing mice, administration of TRi-1 (10 mg/kg, i.v., twice daily for 4 days) did not cause overt toxicity or significant changes in mouse body weight compared to vehicle controls. [1] TRi-1 showed minimal effects on mitochondrial respiration in cell culture, unlike auranofin which caused severe impairment, suggesting a potentially better safety profile regarding mitochondrial toxicity. [1] TRi-1 exhibited reduced cytotoxicity towards various types of non-cancerous primary cells (fibroblasts, keratinocytes, colon epithelial cells) compared to its effects on cancer cell lines. [1] |
| References |
[1]. Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Sci Transl Med. 2018 Feb 14;10(428). pii: eaaf7444. |
| Additional Infomation |
TRi-1 is a selective, irreversible inhibitor of the cytosolic selenoenzyme thioredoxin reductase 1 (TXNRD1). [1] Its proposed mechanism of anticancer action involves two potential components: irreversible inhibition of the antioxidant activity of TXNRD1, and conversion of the inhibited enzyme into pro-oxidant SecTRAPs that generate H2O2, thereby exacerbating oxidative stress in cancer cells. [1] The study proposes that selective inhibition of cytosolic TXNRD1, without targeting the mitochondrial isoform TXNRD2, can yield anticancer efficacy with minimal systemic toxicity, as normal adult tissues may better tolerate loss of TXNRD1 function compared to cancer cells which are addicted to this pathway. [1] TRi-1 was identified through a high-throughput screen of 392,548 compounds. [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~62.5 mg/mL (~190.13 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (6.33 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (6.33 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.0420 mL | 15.2101 mL | 30.4201 mL | |
| 5 mM | 0.6084 mL | 3.0420 mL | 6.0840 mL | |
| 10 mM | 0.3042 mL | 1.5210 mL | 3.0420 mL |