TRPM8 antagonist 2 is a potent and selective TRPM8 antagonist, with an IC50 of 0.2 nM, used in the research of neuropathic pain syndromes.
Physicochemical Properties
| Molecular Formula | C26H26N2O2 |
| Molecular Weight | 398.496846675873 |
| Exact Mass | 398.2 |
| Elemental Analysis | C, 78.36; H, 6.58; N, 7.03; O, 8.03 |
| CAS # | 259674-19-6 |
| PubChem CID | 57055437 |
| Appearance | Light yellow to yellow solid powder |
| LogP | 5.5 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 3 |
| Rotatable Bond Count | 9 |
| Heavy Atom Count | 30 |
| Complexity | 507 |
| Defined Atom Stereocenter Count | 1 |
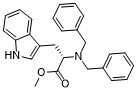
| SMILES | COC(=O)[C@H](CC1=CNC2=CC=CC=C21)N(CC3=CC=CC=C3)CC4=CC=CC=C4 |
| InChi Key | HHVOOJDLCVOLKI-VWLOTQADSA-N |
| InChi Code | InChI=1S/C26H26N2O2/c1-30-26(29)25(16-22-17-27-24-15-9-8-14-23(22)24)28(18-20-10-4-2-5-11-20)19-21-12-6-3-7-13-21/h2-15,17,25,27H,16,18-19H2,1H3/t25-/m0/s1 |
| Chemical Name | methyl (2S)-2-(dibenzylamino)-3-(1H-indol-3-yl)propanoate |
| Synonyms | TRPM8 antagonist 2; HVN74196; HVN-74196; HVN 74196; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | TRPM8 antagonist 2 targets transient receptor potential melastatin 8 (TRPM8) (IC₅₀: 11 nM, determined by FLIPR calcium flux assay; Ki: 7.3 nM, determined by radioligand binding assay) |
| ln Vitro |
TRPM8 Antagonist 2 (Compound 14) is a strong TRPM8 antagonist with an IC50 of 0.2 nM and is employed in the investigation of neuropathic pain syndromes. TRPM8 Antagonist 2 effectively suppresses the menthol-induced increase in intracellular Ca2+ levels in the Ca2+ fluorescence test of HEK293 stable cells expressing the channel subtype of the TRPM8 channel (IC50, 40 nM) [1]. TRPM8 antagonist 2 potently inhibited TRPM8 channel activation induced by menthol (100 μM) or cold mimetic (icilin, 1 μM) in HEK293 cells stably expressing human TRPM8, with an IC₅₀ of 11 nM (FLIPR calcium flux assay) [1] It competitively bound to the TRPM8 receptor with a Ki value of 7.3 nM, as measured by radioligand binding assay using [³H]icilin [1] Exhibited high selectivity for TRPM8, with no significant inhibition (IC₅₀ > 10 μM) of other TRP channels (TRPV1, TRPV3, TRPV4, TRPA1) or ion channels (sodium, potassium channels) tested [1] Did not affect cell viability of HEK293-TRPM8 cells or primary dorsal root ganglion (DRG) neurons at concentrations up to 10 μM (MTT assay) [1] |
| ln Vivo |
Additionally, TRPM8 antagonist 2 (0.1 and 1 μg, subcutaneous injection) can lessen oxaliplatin (OXP)-induced simulated cold allodynia [1]. TRPM8 antagonist 2 (1, 10 and 30 mg/kg, subcutaneous injection) demonstrated significant dose-dependent analgesic activity and inhibited wet dog shock (WDS)-like cold hypersensitivity at 30 mg/kg. In mice cold plate test (5°C), oral administration of TRPM8 antagonist 2 (3, 10, 30 mg/kg) dose-dependently increased the latency to paw licking/flutter, with a maximum effect at 30 mg/kg (latency increased by ~180% vs vehicle control) [1] In the chronic constriction injury (CCI)-induced neuropathic pain model in rats, oral administration of the compound (10, 30 mg/kg, once daily for 7 days) significantly reduced cold allodynia (assessed by acetone drop test) and mechanical hyperalgesia (assessed by von Frey filaments) [1] In the carrageenan-induced inflammatory pain model in mice, oral administration (30 mg/kg) reduced thermal hyperalgesia (hot plate test) and mechanical allodynia, with an efficacy comparable to gabapentin (100 mg/kg) [1] The analgesic effect was rapid in onset (within 1 hour post-administration) and lasted for ~6 hours at the 30 mg/kg dose [1] |
| Enzyme Assay |
FLIPR calcium flux assay: HEK293 cells stably expressing human TRPM8 were seeded in 384-well plates, loaded with a calcium-sensitive fluorescent dye for 1 hour at 37°C [1] Serial dilutions of TRPM8 antagonist 2 (0.1 nM–10 μM) were added, incubated for 30 minutes, then TRPM8 was activated by adding menthol (100 μM) or icilin (1 μM) [1] Fluorescence intensity was measured in real-time using a FLIPR instrument, and IC₅₀ values were calculated by fitting the inhibition curve [1] Radioligand binding assay: Membrane preparations from HEK293-TRPM8 cells were mixed with [³H]icilin (fixed concentration) and serial dilutions of TRPM8 antagonist 2 (0.1 nM–1 μM), incubated at 4°C for 2 hours [1] Bound and free ligand were separated by rapid filtration through glass fiber filters, radioactivity was measured by liquid scintillation counting [1] Non-specific binding was determined in the presence of excess unlabeled icilin, and Ki values were calculated using the Cheng-Prusoff equation [1] |
| Cell Assay |
HEK293 cells stably expressing human TRPM8 were cultured in DMEM medium supplemented with 10% fetal bovine serum and selection antibiotic, maintained at 37°C in a 5% CO₂ incubator [1] Primary DRG neurons were isolated from adult mice, dissociated, and cultured in neurobasal medium supplemented with growth factors for 24–48 hours [1] Cell viability assay: Cells were seeded in 96-well plates, treated with TRPM8 antagonist 2 (0.1 nM–10 μM) for 24 hours, MTT reagent was added, incubated for 4 hours, formazan crystals were dissolved in DMSO, and absorbance was measured at 570 nm [1] Patch-clamp recording (electrophysiology): HEK293-TRPM8 cells were seeded on coverslips, whole-cell patch-clamp recordings were performed at room temperature [1] TRPM8 currents were induced by applying icilin (1 μM) or reducing bath temperature to 15°C, and the effect of TRPM8 antagonist 2 (10, 100 nM) on current amplitude was recorded [1] |
| Animal Protocol |
Male C57BL/6 mice (8–10 weeks old) and Sprague-Dawley rats (200–250 g) were used for in vivo experiments, randomly divided into vehicle control, positive control (gabapentin), and TRPM8 antagonist 2 dose groups (n=8–10 per group) [1] Drug preparation: TRPM8 antagonist 2 was dissolved in 10% DMSO + 90% polyethylene glycol 400 (PEG400) for oral gavage; vehicle control received the same solvent mixture [1] Cold plate test (mice): Mice were placed on a cold plate maintained at 5°C, latency to paw licking/flutter was recorded before and 1, 3, 6 hours after oral administration of TRPM8 antagonist 2 (3, 10, 30 mg/kg) [1] CCI neuropathic pain model (rats): Chronic constriction injury was induced by ligating the sciatic nerve; 7 days post-surgery, rats were treated with TRPM8 antagonist 2 (10, 30 mg/kg) or gabapentin (100 mg/kg) orally once daily for 7 days [1] - Cold allodynia: Acetone drops were applied to the injured paw, and the number of withdrawal responses was counted [1] - Mechanical hyperalgesia: Von Frey filaments of increasing force were applied to the paw, and the paw withdrawal threshold was recorded [1] Carrageenan-induced inflammatory pain model (mice): Carrageenan (1% in saline) was injected into the hind paw; 1 hour later, TRPM8 antagonist 2 (30 mg/kg) or gabapentin (100 mg/kg) was administered orally [1] - Thermal hyperalgesia: Latency to paw withdrawal in the hot plate test (55°C) was recorded [1] - Mechanical allodynia: Paw withdrawal threshold to von Frey filaments was measured [1] |
| ADME/Pharmacokinetics |
In rats, oral administration of TRPM8 antagonist 2 (30 mg/kg) resulted in a bioavailability of 42%, peak plasma concentration (Cmax) of 892 ng/mL, and terminal half-life (t1/2) of 3.7 hours [1] Intravenous administration (10 mg/kg) in rats showed a total clearance (CL) of 18 mL/min/kg and volume of distribution at steady state (Vss) of 0.8 L/kg [1] Exhibited good metabolic stability in human and rat liver microsomes (t1/2 > 60 minutes) [1] Low plasma protein binding (35% in human plasma, 41% in rat plasma) [1] |
| Toxicity/Toxicokinetics |
Acute toxicity study in mice showed no mortality or obvious clinical signs of toxicity (lethargy, loss of appetite, abnormal behavior) at oral doses up to 300 mg/kg [1] Subchronic toxicity study (14 days, oral 30 mg/kg/day) in rats did not cause significant changes in body weight, hematological parameters, or liver/kidney function (ALT, AST, BUN, Cr) [1] |
| References |
[1]. Identification of a Potent Tryptophan-Based TRPM8 Antagonist With in Vivo Analgesic Activity. J Med Chem. 2018 Jul 10. doi: 10.1021/acs.jmedchem.8b00545. |
| Additional Infomation |
TRPM8 antagonist 2 is a potent, selective, tryptophan-based synthetic small-molecule antagonist of TRPM8 [1] Its analgesic mechanism involves blocking TRPM8-mediated cold sensing and nociceptive signaling in DRG neurons [1] It shows potential therapeutic value for the treatment of neuropathic pain (e.g., post-herpetic neuralgia) and inflammatory pain, with favorable pharmacokinetic properties and safety profile [1] The tryptophan scaffold contributes to its high affinity and selectivity for the TRPM8 channel [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~100 mg/mL (~250.94 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.67 mg/mL (6.70 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 26.7 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.67 mg/mL (6.70 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 26.7 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.67 mg/mL (6.70 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 26.7 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5094 mL | 12.5471 mL | 25.0941 mL | |
| 5 mM | 0.5019 mL | 2.5094 mL | 5.0188 mL | |
| 10 mM | 0.2509 mL | 1.2547 mL | 2.5094 mL |