Physicochemical Properties
| Molecular Formula | C20H18CLN3O6 |
| Molecular Weight | 431.826424121857 |
| Exact Mass | 431.088 |
| Elemental Analysis | C, 55.63; H, 4.20; Cl, 8.21; N, 9.73; O, 22.23 |
| CAS # | 1558021-37-6 |
| Related CAS # | 1558021-37-0 |
| PubChem CID | 73052863 |
| Appearance | Light yellow to yellow solid powder |
| LogP | 2.8 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 30 |
| Complexity | 692 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | ClC1C=CC(=CC=1)OC1=CC=C(C=N1)CN1C(C(C(NCC(=O)O)=O)=C(CC1)O)=O |
| InChi Key | HMMHKGLPKAQOOH-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C20H18ClN3O6/c21-13-2-4-14(5-3-13)30-16-6-1-12(9-22-16)11-24-8-7-15(25)18(20(24)29)19(28)23-10-17(26)27/h1-6,9,25H,7-8,10-11H2,(H,23,28)(H,26,27) |
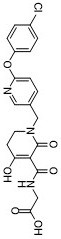
| Chemical Name | 2-[[1-[[6-(4-chlorophenoxy)pyridin-3-yl]methyl]-4-hydroxy-6-oxo-2,3-dihydropyridine-5-carbonyl]amino]acetic acid |
| Synonyms | TP0463518; 1558021-37-6; 2-(1-((6-(4-Chlorophenoxy)pyridin-3-yl)methyl)-4-hydroxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carboxamido)acetic acid; V65GPE6NTB; 2-[[1-[[6-(4-chlorophenoxy)pyridin-3-yl]methyl]-4-hydroxy-6-oxo-2,3-dihydropyridine-5-carbonyl]amino]acetic acid; TP-0463518; (1-((6-(4-Chlorophenoxy)pyridin-3-yl)methyl)-2,4-dioxopiperidine-3-carbonyl)glycine; 2-((1-((6-(4-Chlorophenoxy)pyridin-3-yl)methyl)-4-hydroxy-6-oxo-2,3-dihydropyridine-5-carbonyl)amino)acetic acid; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | HIF-PHD (hypoxia-inducible factor prolyl hydroxylases) (Ki = 5.3 nM) |
| ln Vitro |
Hypoxia-inducible factor prolyl hydroxylases (PHDs) inhibitor stabilizes hypoxia inducible factor alpha, which increases erythropoietin (EPO) expression via the hypoxia response element. Therefore, PHDs inhibitors have been developed as novel therapeutic agents for anemia. Here, we characterize the in vitro and in vivo pharmacological profiles of TP0463518, 2-[[1-[[6-(4-chlorophenoxy)pyridin-3-yl]methyl]-4-hydroxy-6-oxo-2,3-dihydropyridine-5-carbonyl]amino]acetic acid, a novel potent PHDs inhibitor. TP0463518 competitively inhibited human PHD2 with a Ki value of 5.3 nM. TP0463518 also inhibited human PHD1/3 with IC50 values of 18 and 63 nM as well as monkey PHD2 with an IC50 value of 22 nM.
Inhibition of PHDs activity [1] As previously reported, the IC50 values of TP0463518 for human and rat PHD2 were 13 and 18 nM when HIF1α peptide was used as a substrate (Hamada et al., 2018). To elucidate the inhibitory profiles of TP0463518, we evaluated the IC50 values for human PHD1 and PHD3 using HIF1α peptide. TP0463518 inhibited PHD1 with an IC50 value of 18 nM (Table 1). Although TP0463518 also inhibited PHD3, the IC50 values were 3.5 and 4.8 times higher than those of PHD1/2. When using HIF2α peptide as a substrate, TP0463518 inhibited all the PHDs at potencies comparable to those obtained using HIF1α peptide as a substrate. The inhibitory profiles for other species were also investigated. TP0463518 inhibited monkey PHD2 with an IC50 value of 22 nM. To elucidate the mode of inhibition, the maximum activity and Km values were measured at each of the TP0463518 concentrations. The maximum activities (mP values) were 46, 47 and 47 for 0, 20 and 40 nM of TP0463518, respectively. The Km values were 0.10 μM without TP0463518, whereas they were 0.32 and 0.61 μM for 20 and 40 nM of TP0463518, indicating competitive inhibition. Competitive inhibition was confirmed using a double-reciprocal plot (Fig. 2). Based on the competitive inhibition, the Ki value of TP0463518 was calculated as 5.3 nM. |
| ln Vivo |
In normal mice and rats, TP0463518 significantly increased the serum EPO levels at doses of 5 and 20 mg/kg, respectively. The correlation factors for serum EPO and the serum TP0463518 levels were 0.95 in mice and 0.92 in rats. TP0463518 also increased the serum EPO level in 5/6 nephrectomized chronic kidney disease model rats at a dose of 10 mg/kg, with a correlation factor for serum EPO and the serum TP0463518 levels of 0.82. Finally, the effect of TP0463518 in monkeys was investigated. TP0463518 was promptly removed with a half-life of 5.2 h and increased the area under the curve (AUC) of EPO at a dose of 5 mg/kg. The EPO and TP0463518 levels were also correlated. These results suggest that TP0463518 induces endogenous EPO with a strong pharmacokinetic-pharmacodynamic correlation and may contribute to desirable hemoglobin control in patients with renal anemia. [1]
Effect of TP0463518 on serum EPO levels in healthy rodents [1] Next, to elucidate the EPO-producing effect of TP0463518 in rodents, single doses of TP0463518 were administered orally to healthy mice and rats. The serum EPO concentrations in the Balb/c mice at 6 h after administration are shown in Fig. 3A. The serum EPO concentrations increased in a dose-dependent manner, and a significant EPO-producing effect was observed at a dose of 5 mg/kg or more. A pharmacokinetic-pharmacodynamic (PK/PD) analysis showed an excellent correlation between the plasma TP0463518 concentrations and the serum EPO levels with a correlation factor of 0.95 (Fig. 3B). Effect of TP0463518 on serum EPO levels in CKD model rats [1] To study the EPO-producing effect of TP0463518 in a CKD model, TP0463518 was administered to 5/6 Nx rats. As with the SD rats, the serum EPO levels were evaluated at the time when the maximum serum EPO concentration was obtained. The serum EPO levels increased significantly in a dose-dependent manner at a dose of 10 mg/kg or more (Fig. 5A). The serum TP0463518 concentration was also strongly correlated with the serum EPO concentration (Fig. 5B). The serum EPO concentration in the 5/6 Nx rats was comparable to that in the SD rats when the serum TP0463518 concentration was the same (Fig. 4B and Fig. 5B). Effect of TP0463518 on serum EPO levels in monkeys [1] Finally, the effect of TP0463518 was studied in monkeys (Macaca fascicularis). The plasma TP0463518 concentrations peaked at 1.6 h after the administration of 20 mg/kg of TP0463518 and then decreased promptly during the distribution phase (Fig. 6A). The T1/2 during the elimination phase was 5.2 h. The serum EPO concentration peaked at 8 h post-administration in all the dosing groups and then decreased at 24 h (Fig. 6B). The serum EPO AUC increased significantly at a dose of 5 mg/kg or more (Fig. 6C). The serum EPO AUC was correlated with the plasma TP0463518 AUC (Fig. 6D). Prolyl hydroxylase (PHD) 1/2/3 pan inhibitors are known to potentially induce erythropoietin (EPO) production in both the kidney and liver. The 2-[[1-[[6-(4-chlorophenoxy)pyridin-3-yl]methyl]-4-hydroxy-6-oxo-2,3-dihydropyridine-5-carbonyl]amino]acetic acid (TP0463518) is a novel PHD 1/2/3 pan inhibitor; however, the main source of EPO production after TP0463518 administration remained to be investigated. We examined the effect of TP0463518 in inducing EPO production in the kidney and liver by measuring the hypoxia-inducible factor 2α (HIF-2α), EPO mRNA, and serum EPO levels in normal and bilaterally nephrectomized rats. Furthermore, we examined whether liver-derived EPO improved anemia in 5/6 nephrectomized (5/6 Nx) rats. TP0463518 scarcely increased the HIF-2α and EPO mRNA expression levels in the kidney cortex, whereas oral administration of TP0463518 at 40 mg/kg dramatically increased the HIF-2α level from 0.27 to 1.53 fmol/mg and the EPO mRNA expression level by 1300-fold in the livers of healthy rats. After administration of TP0463518 at 20 mg/kg, the total EPO mRNA expression level in the whole liver was 22-fold that in the whole kidney. In bilaterally nephrectomized rats, TP0463518 raised the serum EPO concentration from 0 to 180 pg/ml at 20 mg/kg. Furthermore, repeated administration of TP0463518 at 10 mg/kg increased the reticulocyte count in 5/6 Nx rats on day 7 and raised the hemoglobin level on day 14. The present study revealed that TP0463518 specifically induced EPO production in the liver and improved anemia. The characteristic feature of TP0463518 would lead to not only a more detailed understanding of the PHD-HIF2α-EPO pathway in erythropoiesis, but a new therapeutic alternative for renal anemia. SIGNIFICANCE STATEMENT: Prolyl hydroxylase (PHD) 1/2/3 pan inhibitors are known to potentially induce erythropoietin (EPO) production in both the kidney and liver; however, their effects on renal EPO production have been shown to vary depending on the experimental conditions. The authors found that 2-[[1-[[6-(4-chlorophenoxy)pyridin-3-yl]methyl]-4-hydroxy-6-oxo-2,3-dihydropyridine-5-carbonyl]amino]acetic acid (TP0463518), a PHD 1/2/3 pan inhibitor, specifically induced EPO production in the liver and that the liver-derived EPO was pharmacologically effective. Investigation of the effects of TP0463518 may pave the way for the development of a new therapeutic alternative for renal anemia patients [2]. |
| Enzyme Assay |
Enzymatic assay [1] The PHDs inhibition studies were performed using fluorescence polarization. FITC-HIF and 2-oxoglutarate were mixed with enzyme solution in a reaction buffer (20 mM Tris-HCl [pH 7.5], 5 mM KCl, 1.5 mM MgCl2, 10 μM FeSO4, 2 mM ascorbic acid, 1 mM DTT) with or without various concentrations of TP0463518. The concentrations of FITC-HIF and 2-oxoglutarate were twice the Km values of each enzyme. The reaction temperature was 30 °C, and the reaction time was optimized to each PHD enzyme to obtain the initial velocity (9–20 min). At the end of the reaction, a stop solution containing 20 mM of EDTA and anti-hydroxylated HIF antibody (Cell Signaling Technology, Inc.) was added to the reaction buffer. Then, the fluorescence (ex: 480 nm, em: 535 nm) was measured using EnVision (PerkinElmer Japan Co., Ltd.) to calculate the millipolarization (mP) value. The mP values and the corresponding hydroxylated HIF concentration were proportional, so we used the mP values as the activities. The IC50 values were calculated using SAS version 9.2 (SAS Institute, Tokyo, Japan) using a nonlinear least squares method. To determine the mode of inhibition, the activity of PHD2 was measured with various concentrations of 2-oxoglutarate (0.025–8 μM) and TP0463518 (0–40 μM). Then the apparent Vmax and Km corresponding to each TP0463518 concentration were compared. The mode of inhibition was confirmed using a double reciprocal plot. The Ki value was calculated according to the mode of inhibition (SAS 9.2). |
| Cell Assay |
Determination of serum EPO [1] The serum EPO levels in mice, 5/6 Nx rats, and monkeys were measured using a commercially available EPO ELISA kit according to the manufacturer's manual with slight modifications. The serum EPO levels in healthy rats were measured using a sandwich immunoassay system. The rat EPO concentration from BioLegend and Meso Scale Diagnostics were confirmed to be comparable. EPO levels below the detection limits were calculated as being zero. Determination of plasma/serum TP0463518 concentration [1] The plasma/serum concentrations of TP0463518 were measured using liquid chromatography-tandem mass spectrometry (LC-MS/MS) consisting of an LC‐30AD HPLC syste and a Triple Quad 5500 Mass Spectrometer. |
| Animal Protocol |
Nine-week-old Balb/c mice were randomly assigned to a vehicle or a 5–40 mg/kg dose of TP0463518 group. The mice were orally treated with 0.5% methyl cellulose or a TP0463518 dosing suspension. Blood was collected at 6 h after administration from the orbital plexus under deep anesthesia, and euthanasia was performed without awakening. An aliquot of blood was mixed with EDTA, and the remaining blood sample was left to stand at room temperature for 15 min. The samples were then centrifuged (2130×g for 10 min at 4 °C) to prepare the plasma and serum. For the healthy rats study, 7-week-old SD rats (Japan SLC, Inc.) were randomly assigned to a vehicle or 1.25–160 mg/kg dose of TP0463518 group. For the chronic kidney disease (CKD) model study, 5/6 nephrectomized SD (5/6 Nx) rats were prepared at Japan SLC, Inc., as follows. Two-thirds of the left kidney were resected at 4 weeks of age and the right kidney was removed at 5 weeks of age. The rats were then transferred to our facility and kept until 10 weeks of age, at which time they had developed anemia. The rats were assigned to a vehicle or a 2.5–80 mg/kg dose of TP0463518 group, while ensuring that there was no imbalance in the variance and mean of their whole-blood hemoglobin levels. SD rats and 5/6 Nx rats were orally treated with 0.5% methyl cellulose or a TP0463518 dosing suspension. Approximately 0.6 mL of blood was collected from the tail vein at 8 h (SD rats) or 4 h (5/6 Nx rats) after administration. The serum samples were prepared using the same method as that used for mice. Eight monkeys (9–12-year-old Macaca fascicularis.) were subjected to a fast for 16 h before administration and were re-fed at 8 h post-administration. Blood was collected from the cephalic vein or the femoral vein before (0) and 0.5, 1, 2, 4, 8, 12 and 24 h after administration. Plasma samples were prepared at all the time points, and serum samples were prepared at 0, 4, 8, 12 and 24 h after administration. The experiments were repeated weekly with increasing doses of TP0463518 from 0 (vehicle) to 20 mg/kg. |
| ADME/Pharmacokinetics | The half-life of TP0463518 (T1/2) in monkeys was 5.2 h. This value is very close to the predicted human T1/2 of 1.3–5.6 h estimated from pharmacokinetic parameters obtained in rats and dogs (Hamada et al., 2018). A T1/2 of 5 h might be a sufficient interval, since 2.5 mg/kg, which is one eighth of the effective dose of 20 mg/kg, was ineffective in the monkey study. Based on these results, clinical trials of TP0463518 are now being conducted as a once-daily preparation. Since PHDs inhibitors regulate a wide range of gene expressions, we believe that it is important to address concerns about mechanism-based side effects, especially VEGF induction. In the daprodustat, where the inhibitory activity is close to TP0463518 and the preparation is once-daily, a trend of VEGF was not clearly apparent (Holdstock et al., 2016; Akizawa et al., 2017). To conduct clinical trials in safe, we carefully titrated the dose and monitored VEGF in first-in-human study with healthy volunteers. [1] |
| References |
[1]. TP0463518, a novel inhibitor for hypoxia-inducible factor prolyl hydroxylases, increases erythropoietin in rodents and monkeys with a good pharmacokinetics-pharmacodynamics correlation. Eur J Pharmacol. 2018 Nov 5;838:138-144. [2]. TP0463518, a Novel Prolyl Hydroxylase Inhibitor, Specifically Induces Erythropoietin Production in the Liver. J Pharmacol Exp Ther. 2019 Dec;371(3):675-683. |
| Additional Infomation |
PHD inhibitors protect HIFs alpha from proteasomal degradation by inhibiting HIF alpha hydroxylation (Schmid and Jelkmann, 2016). Subsequently, Epo, which is located downstream of the HIF response element, is upregulated and induces hematopoiesis (Haase, 2006, Percy et al., 2008). Recently, PHD inhibitors have been developed in clinical studies to ameliorate renal anemia, and a series of results showing a clinical proof-of-concept were reported from some companies (Akizawa et al., 2017, Martin et al., 2017, Provenzano et al., 2016). TP0463518 is a glycineamide-type PHDs inhibitor (Hamada et al., 2018) that is presently being examined in a clinical trial. In this report, we summarized the characteristics of TP0463518 in in vitro and in vivo studies. TP0463518 inhibited all human PHD1/2/3 on HIF1α, and it also inhibited rat and monkey PHD2. TP0463518 is a competitive inhibitor to 2-oxoglutarate, and its Ki value for human PHD2 was 5.3 nM. These findings suggest that the potency of TP0463518 is similar to daprodustat which is now in phase 3 trial (Ariazi et al., 2017). The IC50 value of TP0463518 for PHD3 was 3.5 and 4.8 times higher than those for PHD1/2, suggesting that TP0463518 is preferable to PHD1/2. Although TP0463518 had a preference for PHD1/2, the TP0463518 Cmax of monkey was much higher than the IC50 values (i.e., IC50 for human PHD3 63 nM was 27 ng/mL), so TP0463518 was considered to inhibit all the PHDs. TP0463518 also inhibited PHD2 when the substrate was HIF2α. As HIF2α plays an important role in EPO production (Appelhoff et al., 2004, Kapitsinou et al., 2010), the effects of TP0463518 on EPO production were then investigated in in vivo studies. [1] TP0463518 showed a significant EPO-inducing effect in healthy mice and rats from doses of 5 and 20 mg/kg, respectively, with an excellent PK/PD correlation. In renal anemia, EPO production in response to hypoxia is impaired. To investigate the EPO-inducing effect of TP0463518 in renal anemic model animals, 5/6 Nx rats were dosed with TP0463518. TP0463518 induced EPO production with a strong PK/PD correlation. The serum EPO concentration in 5/6 Nx rats was comparable to that in healthy SD rats at the same exposure level. The number of renal EPO-producing (REP) cells in 5/6 Nx rats was estimated to be one sixth of those in healthy SD rats. Therefore, some mechanisms were assumed to increase the serum EPO levels to a certain level after TP0463518 administration regardless of the amount of remnant kidney. The following three possibilities could explain these mechanisms. [1] First, TP0463518 induced an approximately 6-times higher level of EPO production in the remnant damaged kidney in 5/6 Nx rats. When a unilateral ureteral obstruction was created in knockout mice lacking PHD1/2/3 in EPO-producing cells, the EPO mRNA levels in the damaged kidney were reportedly higher than those in the healthy kidney (Souma et al., 2016). In the paper, myofibroblasts, which had been REP cells before transformation, had the potency to express EPO in response to PHD deficiency. So, in our experiment, damaged REP cells in 5/6 Nx rats could have produced more EPO than normal REP cells. [1] A second possibility is that TP0463518 induced more EPO under hypoxic conditions in 5/6 Nx rats. One paper reported that hypoxia and ciclopirox (CPX), which is an iron chelator, synergistically increased reporter gene expressions via EPO HRE (Wanner et al., 2000). Iron chelators deprive iron from the enzyme and seems to inhibit the first step of the reaction (Hoffart et al., 2006). TP0463518 competed with 2-oxoglutarate and also seems to inhibit the first step of the reaction. Therefore, as in the case for CPX, a synergistic effect of TP0463518 and hypoxia might be expected in our experiments. [1] Finally, TP0463518 possibly increased extra-renal EPO production without enhancing EPO production in the kidney. Liver-specific PHD 1/2/3 triple knockout mice are known to have elevated liver EPO production (Minamishima and Kaelin, 2010). TP0463518 is a PHD1/2/3 pan-inhibitor, though the potency is slightly weak in PHD3. In this case, TP0463518 would not have reached REP cells since EPO production in the kidney is up-regulated by single suppression of PHD2 (Takeda et al., 2008). We are now investigating which organ is the main source of EPO and all these possibilities will be examined in future studies. [1] Next, the EPO-producing effect of TP0463518 was investigated in monkeys (Macaca fascicularis). The serum EPO AUC was correlated with the plasma TP0463518 AUC and increased significantly at a dose of 5 mg/kg or more. The EPO AUC, and not the EPO Cmax, is important for increasing blood hemoglobin levels (Masunaga et al., 1989). Because high levels of hemoglobin increase the risks of cardiovascular disease and stroke (Pfeffer et al., 2009, Singh et al., 2006), controlling the EPO AUC is very important for maintaining adequate levels of hemoglobin. Unlike exogenous erythropoiesis stimulating agent, as PHD inhibitor regulates endogenous EPO levels, a strong PK/PD correlation would lead to desirable hemoglobin control. A previous report suggested that a high dose of recombinant EPO used to treat anemia patients, which greatly exceeded normal physiologic ranges of EPO, might increase the risk of a cardiovascular event independent to blood pressure rise (Szczech et al., 2008, Inrig et al., 2012). In our experiment in monkeys, the serum EPO increased to 60 mU/mL at a dose of 20 mg/kg. This increase is comparable to the physiologic increase in endogenous EPO observed at high altitudes (Klausen et al., 1996) and is sufficient to ameliorate anemia when administered once daily in not only monkeys but also human (Akizawa et al., 2017, Flamme et al., 2014, Holdstock et al., 2016). As Flamme et al. discussed, erythropoiesis stimulating agents therapy, which leads serum EPO concentration over normal physiologic range, has a potential of long-term safety concern and a therapy with PHD inhibitors might not need such a high exposure of EPO. Therefore, TP0463518, which induced effective levels but not excess normal physiologic ranges of EPO, could ameliorate anemia with a lower risk of cardiovascular events that observed for recombinant EPO. [1] The systemic conditional knockout of PHD2 increases the serum VEGF concentration (Takeda et al., 2007). To reduce mechanism-based adverse effects, an interval during which the PHDs inhibitor does not work is considered important. The half-life of TP0463518 (T1/2) in monkeys was 5.2 h. This value is very close to the predicted human T1/2 of 1.3–5.6 h estimated from pharmacokinetic parameters obtained in rats and dogs (Hamada et al., 2018). A T1/2 of 5 h might be a sufficient interval, since 2.5 mg/kg, which is one eighth of the effective dose of 20 mg/kg, was ineffective in the monkey study. Based on these results, clinical trials of TP0463518 are now being conducted as a once-daily preparation. Since PHDs inhibitors regulate a wide range of gene expressions, we believe that it is important to address concerns about mechanism-based side effects, especially VEGF induction. In the daprodustat, where the inhibitory activity is close to TP0463518 and the preparation is once-daily, a trend of VEGF was not clearly apparent (Holdstock et al., 2016; Akizawa et al., 2017). To conduct clinical trials in safe, we carefully titrated the dose and monitored VEGF in first-in-human study with healthy volunteers. [1] Hypertension is a well-known adverse event observed in erythropoiesis therapy. Since TP0463518 induced EPO not exceeding the normal physiologic range, we believe that the risk of hypertension is low. Actually, there were no observed trends in blood pressure in phase 2 clinical study in vadadustat, which induces EPO not exceeding the normal physiologic range (Martin et al., 2017, Pergola et al., 2016), and hypertension was only observed in few patients in daprodustat, whose potency is close to TP0463518 (Akizawa et al., 2017). It will be soon reported that TP0463518 does not affect vital signs including blood pressure after single administration (Shinfuku et al., 2018). Based on these information, we believe that the risk of hypertension is low in TP0463518. Nevertheless, we plan to carefully monitor blood pressure in future clinical trials. [1] In summary, TP0463518 competitively inhibited human PHDs and also inhibited rat and monkey PHD2. TP0463518 increased serum EPO levels not only in healthy rodents, but also in anemic rats and monkeys. The serum EPO concentrations were well correlated with TP0463518 exposure in all the animals tested. TP0463518 is now being examined in a clinical trial with a once-daily dose regimen, and a clinical proof-of-concept for TP0463518 will be available in the future. TP0463518 is expected to become a new therapeutic option for the easy control of hemoglobin levels in renal anemia patients. |
Solubility Data
| Solubility (In Vitro) | DMSO : ≥ 125 mg/mL (~289.47 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (4.82 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (4.82 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (4.82 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3157 mL | 11.5786 mL | 23.1573 mL | |
| 5 mM | 0.4631 mL | 2.3157 mL | 4.6315 mL | |
| 10 mM | 0.2316 mL | 1.1579 mL | 2.3157 mL |