Physicochemical Properties
| Molecular Formula | C11H11CL3N4 |
| Molecular Weight | 305.5908 |
| Exact Mass | 304.004 |
| CAS # | 1638211-05-8 |
| Related CAS # | TH287;1609960-30-6 |
| PubChem CID | 91826506 |
| Appearance | Typically exists as solid at room temperature |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 4 |
| Rotatable Bond Count | 2 |
| Heavy Atom Count | 18 |
| Complexity | 254 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | ClC1C(=C([H])C([H])=C([H])C=1C1=C([H])C(=NC(N([H])[H])=N1)N([H])C([H])([H])[H])Cl.Cl[H] |
| InChi Key | YBLIJWDQSUDCJU-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C11H10Cl2N4.ClH/c1-15-9-5-8(16-11(14)17-9)6-3-2-4-7(12)10(6)13;/h2-5H,1H3,(H3,14,15,16,17);1H |
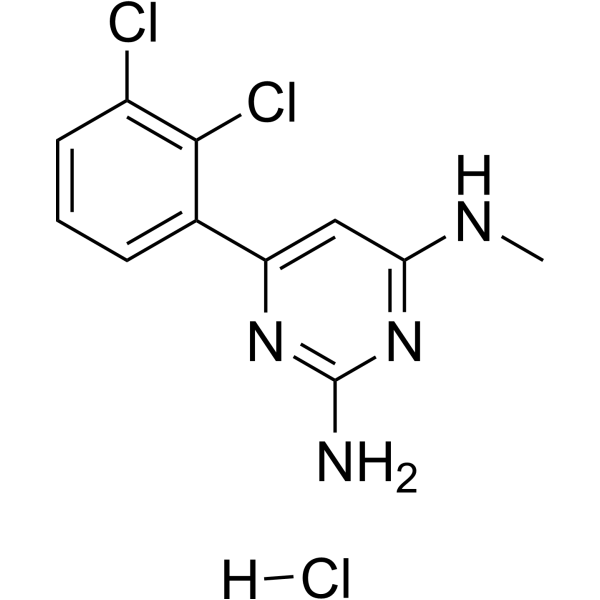
| Chemical Name | 6-(2,3-dichlorophenyl)-4-N-methylpyrimidine-2,4-diamine;hydrochloride |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | While TH287 (1–10 μM; 24 hours) is significantly less toxic to some primary or immortalized cells, it kills U2OS and other cancer cell lines with efficiency and selectivity [1]. |
| ln Vitro |
While TH287 (1–10 μM; 24 hours) is significantly less toxic to some primary or immortalized cells, it kills U2OS and other cancer cell lines with efficiency and selectivity [1]. TH287 potently inhibited MTH1 enzymatic activity in a biochemical assay using dGTP as substrate (IC50 = 0.8 ± 0.1 nM). The inhibition was also effective when using the physiological substrates 8-oxodGTP (IC50 = 0.9 ± 0.2 nM) and 2-OH-dATP (IC50 = 2.5 ± 0.1 nM).[1] In a cellular thermal shift assay (CETSA), TH287 (50 µM) was shown to engage and stabilize the MTH1 protein in intact BJ SV40T RasV12 cells, indicating target engagement.[1] TH287 selectively and effectively killed various cancer cell lines (e.g., U2OS, HeLa, SW620, MDA-MB-231, MCF-7) in viability and clonogenic survival assays, with IC50 values typically in the low micromolar range (e.g., 0.8-1.7 µM for several lines). In contrast, it was considerably less toxic to several primary or immortalized non-transformed cells (e.g., VH10, HDFn, BJ-hTERT), with IC50 values around 20 µM.[1] Treatment of cancer cells (U2OS) with TH287 (10 µM) for 24 hours increased the incorporation of oxidized nucleotides, specifically 8-oxodG and 2-OH-dA, into DNA, as detected by modified comet assays using the repair enzymes OGG1 and MUTYH. This effect was not observed in primary VH10 cells or with the inactive analog TH650.[1] TH287 induced DNA damage in cancer cells, as evidenced by increased formation of 53BP1, RPA, and phospho-DNA-PKcs foci, and triggered an ATM-p53-mediated DNA damage response (phosphorylation of ATM and p53, induction of p21).[1] TH287 induced apoptosis in U2OS cells, as shown by increased caspase-3 cleavage and sub-G1 population in FACS analysis.[1] The cytotoxicity of TH287 was dependent on MTH1 inhibition, as overexpression of wild-type, but not catalytically dead (E56A), MTH1 protein rescued cell viability. Furthermore, expression of MTH1 mutants defective in specific substrate hydrolysis (D119A for 8-oxodGTP, W117Y for 2-OH-dATP) did not rescue viability, indicating both oxidized nucleotide types contribute to toxicity.[1] The cytotoxicity of TH287 was independent of p53 status, as p53-depleted or p53-null cell lines (e.g., HCT116 p53-/-) showed similar sensitivity.[1] Overexpression of the DNA repair enzymes OGG1 or MUTYH did not alter the cytotoxic effect of TH287.[1] In a stepwise transformation model using BJ fibroblasts, TH287 was selectively cytotoxic to cells expressing SV40 large T antigen and oncogenic Ras (BJ-SV40-Ras), but not to parental or hTERT-immortalized (BJ-hTERT) cells.[1] Expression of the bacterial homolog MutT (which is not inhibited by TH287) in the nucleus or mitochondria of human cells partially rescued the cytotoxicity and reduced 53BP1 foci formation induced by TH287, providing strong evidence for on-target mechanism.[1] |
| ln Vivo |
In mice, TH287 (5 mg/kg; ip) has a tmax of 0.5 hours and a Cmax of 0.82 μM [2]. In a mouse xenograft model using SW480 colorectal cancer cells, daily subcutaneous administration of TH287 (the specific dose for TH287 in this model is not provided; the related compound TH588 was used at 25 mg/kg in a similar study) inhibited tumor growth.[1] |
| Enzyme Assay |
A malachite green-based enzymatic assay was used to screen for and characterize MTH1 inhibitors. Purified recombinant human MTH1 protein was incubated with dGTP substrate in an assay buffer. An excess of inorganic pyrophosphatase was added to convert the released pyrophosphate (PPi) into inorganic phosphate (Pi), which was then quantified colorimetrically using malachite green reagent. For IC50 determination, TH287 was serially diluted in DMSO and then in assay buffer. MTH1, dGTP, and pyrophosphatase were added, and the reaction mixture was incubated. The reaction was stopped, and the malachite green reagent was added. After color development, absorbance was read at 630 nm. Dose-response curves were fitted to calculate IC50 values.[1] A more sensitive luminescence-based assay (PPiLight Inorganic Pyrophosphate Assay) was used with the physiological substrates 8-oxodGTP and 2-OH-dATP. MTH1 was incubated with the substrate at its Km concentration in reaction buffer in the presence of serially diluted TH287. After the reaction, released PPi was detected using the commercial kit, and luminescence was measured.[1] Isothermal Titration Calorimetry (ITC) was used to confirm direct binding. TH287 was titrated into a solution containing MTH1 protein. The heat change upon binding was measured, and data were fitted to a binding model to obtain the association constant (K), enthalpy change (ΔH), and stoichiometry (n).[1] Surface Plasmon Resonance (SPR) analysis was performed using a Biacore T200 instrument. Purified MTH1 was immobilized on a CM5 chip. TH287 at various concentrations was flowed over the chip in running buffer. Sensorgrams were recorded, and data were analyzed to determine kinetic parameters (association/dissociation rates) and affinity (Kd).[1] |
| Cell Assay |
For clonogenic survival assays, cells were seeded at low density (e.g., 500 cells per 10-cm dish or 200 cells per well in 6-well plates). After attachment, cells were treated with TH287 or vehicle (DMSO) for 7-14 days. The medium containing the compound was not refreshed during this period. Plates were then fixed and stained with methylene blue, and colonies were manually counted. Survival was normalized to vehicle-treated controls.[1] For cell viability assays (Resazurin/Alamar Blue), cells were seeded in 96-well plates (1,500-3,000 cells per well). The next day, cells were treated with a range of TH287 concentrations. After 72 hours, resazurin solution was added to the wells. After incubation (e.g., 2-4 hours), fluorescence (Ex/Em 530/590 nm or 560/590 nm) was measured. Viability was calculated relative to vehicle-treated controls, and dose-response curves were generated to determine IC50 values.[1] For immunofluorescence (IF) analysis of DNA damage foci, cells were seeded on coverslips or in 96-well plates. After treatment with TH287, cells were fixed with paraformaldehyde, permeabilized, blocked, and incubated with primary antibodies (e.g., against 53BP1, RPA, phospho-DNA-PKcs). After washing, cells were incubated with fluorescently labeled secondary antibodies and DNA counterstain (DAPI or To-Pro-3). Images were acquired by confocal microscopy or high-content imaging, and foci were quantified manually or automatically.[1] For the detection of 8-oxodG incorporation into DNA, cells were fixed with methanol. After permeabilization and blocking, cells were incubated with Alexa Fluor 488-conjugated avidin, which binds specifically to 8-oxodG. DNA was counterstained, and fluorescence intensity in the nucleus was quantified by microscopy or high-content analysis.[1] For the modified comet assay to detect specific oxidized bases, cells treated with TH287 were embedded in low-melting agarose on slides. Cells were lysed, and slides were washed. The DNA was then incubated with buffer alone, OGG1 enzyme (to cut at 8-oxodG sites), or MUTYH enzyme (to cut at 8-oxodG and 2-OH-dA sites). Slides were subjected to alkaline electrophoresis, stained with a DNA dye (YOYO-1), and imaged. DNA damage (tail moment) was quantified using comet analysis software.[1] For apoptosis analysis by FACS, cells treated with TH287 were harvested, fixed, permeabilized, and stained with an antibody against cleaved caspase-3 followed by a fluorescent secondary antibody. Cells were also stained with propidium iodide (PI) and RNase. Samples were analyzed by flow cytometry to quantify the percentage of cleaved caspase-3 positive cells and the sub-G1 (apoptotic) population in the PI histogram.[1] For siRNA transfection, cells were seeded and transfected the next day with siRNA duplexes targeting MTH1 or a non-targeting control using a commercial transfection reagent according to the manufacturer's protocol. Cells were typically analyzed 3-6 days post-transfection for viability, protein expression (western blot), or DNA damage markers (IF).[1] |
| Animal Protocol |
For pharmacokinetic (PK) studies, C57BL/6 or SCID mice (female, 6-8 weeks old) received a single subcutaneous (s.c.) injection of TH287 at 10 mg/kg. Blood samples were collected at various time points post-injection from groups of mice (e.g., n=3 per time point). Plasma was separated and analyzed by LC-MS/MS to determine compound concentration over time.[1] (The specific animal efficacy protocol for TH287 alone is not detailed separately from TH588. The article describes efficacy studies primarily with TH588. A protocol for a related xenograft study involved subcutaneous inoculation of cancer cells (e.g., SW480) into SCID mice. When tumors became palpable, mice were randomized and treated with vehicle or compound via subcutaneous injection once daily. Tumor volumes and body weights were monitored regularly.)[1] |
| ADME/Pharmacokinetics |
In an in vitro metabolic stability assay using mouse liver microsomes (MLM), TH287 showed high intrinsic clearance (CLint = 261 µL/min/mg protein), indicating rapid metabolism.[1] In an in vitro plasma protein binding assay using rapid equilibrium dialysis (RED), TH287 was highly protein bound, with an unbound fraction (fu) of 8.0% in human plasma.[1] The solubility of TH287 in PBS was determined to be 52 µM.[1] In a Caco-2 permeability assay, the apparent permeability (Papp) of TH287 in the apical-to-basolateral (A-B) direction was 8.4 × 10⁻⁶ cm/s, and the efflux ratio (B-A/A-B) was 0.8, suggesting low to moderate permeability and no significant efflux.[1] In vivo, following a single 10 mg/kg subcutaneous dose in mice, TH287 reached a maximum plasma concentration (Cmax) of 1.5 µM at 1 hour (Tmax). The area under the concentration-time curve from 0 to infinity (AUC0-inf) was 1.8 µmol·h/L. The terminal half-life (t1/2) was 0.4 hours. A major metabolite (presumably the N-dealkylated product) was detected with higher exposure (Cmax 5.6 µM, AUC0-inf 10.5 µmol·h/L) and a longer half-life (1.0 h).[1] |
| Toxicity/Toxicokinetics |
In the in vivo single-dose PK study in mice, no acute toxicity observations were reported at the 10 mg/kg subcutaneous dose.[1] (The main article text does not report specific in vivo toxicity parameters for TH287 such as maximum tolerated dose (MTD), lethal dose (LD50), or detailed histopathological analysis of organs.)[1] In vitro, TH287 showed significantly lower cytotoxicity in non-transformed primary or immortalized cells compared to cancer cells, suggesting a potential therapeutic window.[1] |
| References |
[1]. Gad H, et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature. 2014 Apr 10;508(7495):215-21. [2]. Saleh A, et, al. Development and validation of method for TH588 and TH287, potent MTH1 inhibitors and new anti-cancer agents, for pharmacokinetic studies in mice plasma. J Pharm Biomed Anal. 2015 Feb;104:1-11. |
| Additional Infomation |
TH287 was identified as a first-in-class inhibitor of the nudix hydrolase protein family, specifically targeting MTH1. Its discovery stemmed from the concept of non-oncogene addiction, where cancer cells with dysfunctional redox regulation and elevated ROS become dependent on MTH1 to sanitize the oxidized dNTP pool and prevent lethal DNA damage upon replication.[1] The mechanism of action involves inhibition of MTH1's catalytic activity, leading to the incorporation of oxidized dNTPs (like 8-oxodGTP and 2-OH-dATP) into DNA during replication. This incorporation causes DNA damage, activates the DNA damage response, and ultimately leads to cancer cell death, while sparing normal cells with lower oxidative stress.[1] TH287 was found to be rapidly metabolized in vivo via N-dealkylation of its aminomethyl substituent, which led to the development of the more metabolically stable analog TH588 with a cyclopropyl group replacement.[1] The co-crystal structure of MTH1 in complex with TH287 was solved to 1.6 Å resolution, revealing that the aminopyrimidine moiety binds in the active site, forming key hydrogen bonds with Asn33, Asp119, and Asp120.[1] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2724 mL | 16.3618 mL | 32.7236 mL | |
| 5 mM | 0.6545 mL | 3.2724 mL | 6.5447 mL | |
| 10 mM | 0.3272 mL | 1.6362 mL | 3.2724 mL |