TGX-221 is a novel, potent, selective, and cell membrane permeable inhibitor of the PI3K p110β catalytic subunit with potential anticancer activity. TGX221 selectively inhibits renal cell carcinoma cells that have both VHL and SETD2 mutations and connects various pathways. In nude mice, the prostate cancer xenograft tumor is inhibited by TGX221. ccRCC with both VHL and SETD2 mutations can be inhibited specifically by TGX221. Cancer cells with PTEN and CDKN2A mutations were also the target of TGX221. Inhibiting cell motility and tumorigenesis in ccRCC cells with VHL and SETD2 mutations was another area in which TGX221 demonstrated notable selectivity. For ccRCC with VHL and SETD2 mutations, the novel inhibitor TGX221 has a high level of selectivity.
Physicochemical Properties
| Molecular Formula | C21H24N4O2 |
| Molecular Weight | 364.4409 |
| Exact Mass | 364.189 |
| Elemental Analysis | C, 69.21; H, 6.64; N, 15.37; O, 8.78 |
| CAS # | 663619-89-4 |
| Related CAS # | 663619-89-4 |
| PubChem CID | 9907093 |
| Appearance | Off-white to pale yellow |
| Density | 1.3±0.1 g/cm3 |
| Boiling Point | 557.3±60.0 °C at 760 mmHg |
| Flash Point | 290.8±32.9 °C |
| Vapour Pressure | 0.0±1.5 mmHg at 25°C |
| Index of Refraction | 1.646 |
| LogP | 3.19 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 27 |
| Complexity | 710 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | O1C([H])([H])C([H])([H])N(C2=C([H])C(N3C([H])=C(C([H])([H])[H])C([H])=C(C3=N2)C([H])(C([H])([H])[H])N([H])C2C([H])=C([H])C([H])=C([H])C=2[H])=O)C([H])([H])C1([H])[H] |
| InChi Key | CPRAGQJXBLMUEL-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C21H24N4O2/c1-15-12-18(16(2)22-17-6-4-3-5-7-17)21-23-19(13-20(26)25(21)14-15)24-8-10-27-11-9-24/h3-7,12-14,16,22H,8-11H2,1-2H3 |
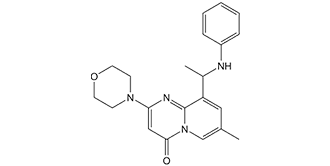
| Chemical Name | 9-(1-anilinoethyl)-7-methyl-2-morpholin-4-ylpyrido[1,2-a]pyrimidin-4-one |
| Synonyms | TGX221; TGX 221; TGX221 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
p110β (IC50 = 8.5 nM); p110δ (IC50 = 211 nM) Phosphatidylinositol 3-Kinase β (PI3Kβ) - IC50 ~3 nM (recombinant human PI3Kβ, HTRF kinase activity assay); - High selectivity over other PI3K subtypes: IC50 > 1000 nM (PI3Kα), >500 nM (PI3Kγ), >800 nM (PI3Kδ) (same assay as PI3Kβ); - No significant inhibition of 30+ unrelated kinases (e.g., AKT, MAPK, PKC) at 1 μM[1] |
| ln Vitro |
The activity of TGX-221 against different isoforms is measured in an in vitro PI3K assay using multiple preparations of recombinant p85/p110. With an IC50 of 211 nM, TGX-221 exhibits slow potency to p110. Additionally, TGX-221 partially inhibits the phosphorylation of Ser473 of PKB that insulin causes in J774.2 macrophage cells. [1] A platelet-ECC interaction, platelet aggregation, and platelet-granulocyte binding model using extracorporeal circulation (ECC) is inhibited by TGX-221.[2] A recent study demonstrates that PC3 cells exhibit inhibition of proliferation following treatment with TGX-221 (0.2, 2, and 20 μM), along with a notable decrease in the activity of the p110β PI3K isoform. [3] 1. PI3Kβ inhibition and signaling suppression (Literature [1]): - Recombinant PI3Kβ activity: TGX-221 (0.1-100 nM) dose-dependently inhibited PI3Kβ; 3 nM inhibited activity by ~50% (IC50), 30 nM by ~90%. No effect on PI3Kα/γ/δ (<10% inhibition at 100 nM). - HEK293 cells overexpressing PI3Kβ: 10 nM TGX-221 reduced insulin-induced p-AKT (Ser473) by ~75% (Western blot) at 30 minutes; 50 nM reduced p-AKT by ~90%. No effect on p-ERK (MAPK pathway) or p-PKC, confirming pathway specificity. - Primary rat aortic smooth muscle cells: 50 nM TGX-221 inhibited PDGF-induced proliferation by ~60% (³H-thymidine incorporation) at 48 hours; cell viability remained >90% (MTT assay)[1] 2. Platelet function inhibition (Literature [2]): - Human platelets (isolated from healthy donors): TGX-221 (1-100 nM) dose-dependently inhibited ADP-induced platelet aggregation. IC50 ~10 nM; 100 nM reduced aggregation by ~95% (light transmission aggregometry). - Mouse platelets: 50 nM TGX-221 inhibited collagen-induced platelet adhesion to fibrinogen by ~80% (static adhesion assay); reduced platelet-derived growth factor (PDGF) release by ~70% (ELISA)[2] |
| ln Vivo |
In a mouse model, integrated blood flow is improved over 30 minutes by TGX-221 at doses 1 + 1 (49%) and 3 + 3 (88%) as an anti-thrombotic agent. In addition, TGX-221 doses of 3 + 3 (median 1560) and 1 + 1 (1305) cause an increase in tail bleeding time(BT) (sec), and all TGX-221 groups experience an increase in mean renal BT (sec).[4] 1. Mouse antithrombotic activity (Literature [2]): - Arterial thrombosis model (ferric chloride-induced carotid artery thrombosis): - Animals: Male C57BL/6 mice (8-10 weeks old). - Administration: TGX-221 dissolved in 10% DMSO + 90% saline, intravenous (i.v.) injection 0.5, 1, 2 mg/kg 15 minutes before thrombosis induction. - Efficacy: 2 mg/kg TGX-221 prolonged time to occlusion from ~25 minutes (vehicle) to ~70 minutes; reduced thrombus weight by ~65% (vs. vehicle). - Tail bleeding time assay: 2 mg/kg i.v. TGX-221 prolonged bleeding time from ~2 minutes (vehicle) to ~6 minutes; no excessive bleeding (bleeding stopped within 10 minutes)[2] |
| Enzyme Assay |
IC50 values are measured using a standard lipid kinase activity with PI as a substrate. (i)100 μM cold ATP is used instead of 10 μM, (ii) 1% DMSO is used, and (iii) [γ-33P]ATP is used instead of [γ-32P]ATP. A phosphorimager screen is used to quantify the TLC plates. The reported IC50 values were calculated using non-linear regression analysis based on at least three independent experiments that were carried out repeatedly using different recombinant protein preparations. 1. Reagent preparation: - Recombinant human PI3Kβ (catalytic subunit p110β + regulatory subunit p85α) resuspended in assay buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT, 0.01% Tween 20). - Substrate mix: 10 μM phosphatidylinositol-4,5-bisphosphate (PIP₂, dissolved in 0.1% CHAPS) + 2 μM ATP + 0.1 μCi [γ-³³P]-ATP dissolved in assay buffer[1] 2. Assay setup: 50 μL reaction mixture contained 5 nM PI3Kβ, substrate mix, and serial concentrations of TGX-221 (0.01-1000 nM). Vehicle control (0.1% DMSO) was included. The mixture was incubated at 30℃ for 60 minutes to allow kinase reaction (PIP₂ phosphorylation to PIP₃)[1] 3. Product detection: - Reaction terminated by adding 100 μL ice-cold 5% trichloroacetic acid (TCA) to precipitate proteins. Samples were centrifuged at 12,000×g for 10 minutes at 4℃; supernatants were collected. - Supernatants were spotted onto polyethyleneimine-cellulose TLC plates, which were developed with chloroform/methanol/water/ammonia (65:25:4:1, v/v/v/v) to separate [³³P]-PIP₃ (product) from [³³P]-ATP (unreacted substrate). - TLC plates were exposed to autoradiography film; radioactivity of [³³P]-PIP₃ spots was quantified via phosphorimager. - Inhibition rate = (1 - radioactivitydrug/radioactivityvehicle) × 100%. IC50 was calculated by nonlinear regression of the concentration-inhibition curve[1] [1] |
| Cell Assay |
Cells are plated in triplicate in 96-well culture plates and allowed to attach for the duration of the incubation period in order to measure cell proliferation. A total of 24, 48, and 72 hours are spent incubating the cells with TGX-221. A crystal violet staining-based colorimetric assay is used to quantify cells at predetermined time intervals. In a nutshell, 100 μl of 2.5% glutaraldehyde solution is added, and 30 minutes are spent incubating the cells at room temperature. Plates are immersed in PBS solution three times to wash them. Prior to binding dye solubilization in 100 μL of 10% acetic acid, plates are air-dried and stained by adding 100 L of 0.1% solution of crystal violet dissolved in deionized water and incubating for 20 minutes at room temperature. Excess dye is removed by extensive washing with deionized water. A microplate reader operating at 570 nm is used to directly measure the optical density of dye extracts in plates. 1. HEK293 cell signaling assay (Literature [1]): - Cell culture: HEK293 cells stably transfected with human PI3Kβ were maintained in DMEM + 10% FBS, seeded in 6-well plates (2×10⁵ cells/well) and cultured overnight at 37℃, 5% CO₂. - Treatment: Cells were serum-starved for 16 hours, then pre-incubated with TGX-221 (1-100 nM) for 1 hour. Insulin (100 nM) was added to stimulate PI3K-AKT signaling for 30 minutes. - Detection: Cells were lysed with RIPA buffer (containing protease/phosphatase inhibitors). Proteins were separated by SDS-PAGE, transferred to PVDF membranes, and probed with antibodies against p-AKT (Ser473), total AKT, p-ERK, and total ERK (loading control). Band intensity was quantified via ImageJ[1] 2. Platelet aggregation assay (Literature [2]): - Platelet isolation: Human peripheral blood was collected into citrate-anticoagulant tubes, centrifuged at 150×g for 15 minutes to obtain platelet-rich plasma (PRP). PRP was adjusted to 2×10⁸ platelets/mL with platelet-poor plasma (PPP). - Treatment: PRP was pre-incubated with TGX-221 (1-100 nM) for 5 minutes at 37℃. ADP (10 μM) was added to induce aggregation. - Detection: Platelet aggregation was monitored for 10 minutes via light transmission aggregometry (LTA), with PPP as 100% transmission and PRP as 0% transmission. Aggregation rate was calculated as the percentage of maximum light transmission[2] [1][2] |
| Animal Protocol |
Rats are randomly assigned to treatment groups that contain the drug wortmannin (an irreversible non-specific PI3K inhibitor), the vehicle propylene glycol (0.25 mL/kg), LY294002 (2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one; a reversible non-specific PI3K inhibitor), and IC87114 (2-[(6-aminopurin- 9-yPI3K p110 antagonist 5-methyl-3-(2-methylphenyl)quinazolin-4-one (2.5 mg/kg) and the selective PI3K p110β antagonist TGX221 (2.5 mg/kg). In the tail bleeding experiments, rats are randomLy assigned to drug treatment groups consisting of LY294002 (10 mg/kg), IC87114 (2.5 mg/kg), wortmannin (5 mg/kg), TGX221 (2.5 or 25 mg/kg), heparin (100 U/kg), aspirin (2× 200 mg/kg p.o.)± heparin (100 U/kg), and aspirin (2× 200 mg/kg p.o.) combined with heparin (100 U/kg) and TGX221 (2.5 mg/kg). All medications—aside from aspirin—are given as a slow (over ≈45-60 s) intravenous bolus into the jugular vein. Two oral doses of aspirin (200 mg/kg suspended in 15% gum arabic in water) are given, the first 24 hours before the experiment and the second one hour before it begins. 1. Animals: Male C57BL/6 mice (8-10 weeks old), 6 mice per group; acclimated to the laboratory for 7 days (12-hour light/dark cycle, ad libitum food and water)[2] 2. Drug preparation: TGX-221 was dissolved in a mixture of 10% DMSO and 90% sterile saline (v/v), sonicated for 5 minutes to ensure complete dissolution. Doses of 0.5, 1, and 2 mg/kg were prepared by adjusting the drug concentration[2] 3. Arterial thrombosis induction and assessment: - Mice were anesthetized with ketamine/xylazine (100/10 mg/kg, i.p.). The right carotid artery was exposed, and a 2×2 mm piece of filter paper soaked in 10% ferric chloride was applied to the artery to induce thrombosis. - TGX-221 (0.5/1/2 mg/kg) or vehicle (10% DMSO + 90% saline) was injected via the tail vein 15 minutes before ferric chloride application. - Blood flow in the carotid artery was monitored using a Doppler flowmeter. Time to complete occlusion (no detectable blood flow for 10 consecutive minutes) was recorded. After occlusion, the thrombus was dissected, blotted dry, and weighed[2] 4. Tail bleeding time assay: - Mice were anesthetized as above. The distal 5 mm of the tail was amputated with a scalpel, and the tail was immediately immersed in 37℃ saline. - Bleeding time was defined as the time from amputation to complete cessation of bleeding (no visible blood for 30 seconds). A maximum cutoff time of 15 minutes was set to avoid excessive blood loss[2] |
| Toxicity/Toxicokinetics |
1. In vitro toxicity:
- HEK293 cells, rat aortic smooth muscle cells, and human platelets: TGX-221 concentrations up to 1 μM showed no non-specific cytotoxicity. For platelets, no morphological changes (e.g., activation-related shape change) were observed at 100 nM (light microscopy); for adherent cells, trypan blue exclusion assay showed >90% viability after 48-hour exposure to 50 nM TGX-221[1] [2] 2. In vivo toxicity: - Mice injected with TGX-221 (0.5-2 mg/kg, i.v.) showed no mortality or abnormal behavior (e.g., ataxia, lethargy) within 24 hours. No significant changes in body weight or gross organ morphology (liver, kidney, spleen) were observed at euthanasia[2] |
| References |
[1]. Biochem J. 2007 Jun 15;404(3):449-58. [2]. Thromb Haemost. 2008 Mar;99(3):609-15. [3]. Appl Microbiol Biotechnol. 2011 Mar;89(5):1423-33. |
| Additional Infomation |
9-(1-anilinoethyl)-7-methyl-2-(4-morpholinyl)-4-pyrido[1,2-a]pyrimidinone is a pyridopyrimidine. 1. Mechanism of action: TGX-221 is a selective PI3Kβ inhibitor that binds to the ATP-binding pocket of the p110β catalytic subunit of PI3Kβ. This binding blocks PI3Kβ-mediated phosphorylation of PIP₂ to PIP₃, thereby inhibiting downstream activation of the AKT signaling pathway. In platelets, this suppresses aggregation and adhesion; in vascular smooth muscle cells, it inhibits proliferation induced by growth factors (e.g., PDGF)[1] [2] 2. Preclinical significance: - Literature [1]: Establishes TGX-221 as a tool compound for studying PI3Kβ-specific functions, particularly in vascular cell signaling and proliferation[1] - Literature [2]: Demonstrates the potential of PI3Kβ inhibitors (e.g., TGX-221) as antithrombotic agents, with efficacy in reducing arterial thrombosis without causing severe bleeding—addressing the limitation of traditional anticoagulants (high bleeding risk)[2] 3. Limitations: - No clinical development data (e.g., FDA status) reported; TGX-221 is primarily used as a preclinical research tool rather than a therapeutic candidate. Literature [3] does not provide any relevant data on TGX-221[1] [2][3] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~12 mg/mL (~32.9 mM) Water: <1 mg/mL (slightly soluble or insoluble) Ethanol: <1 mg/mL (slightly soluble or insoluble) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 1.25 mg/mL (3.43 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 12.5 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 1.25 mg/mL (3.43 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 12.5 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 1.25 mg/mL (3.43 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 12.5 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 1%DMSO+30% polyethylene glycol+1%Tween 80: 30mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7439 mL | 13.7197 mL | 27.4394 mL | |
| 5 mM | 0.5488 mL | 2.7439 mL | 5.4879 mL | |
| 10 mM | 0.2744 mL | 1.3720 mL | 2.7439 mL |