Physicochemical Properties
| Molecular Formula | C27H18F4N4O3S | |
| Molecular Weight | 554.52 | |
| Exact Mass | 554.103 | |
| Elemental Analysis | C, 58.48; H, 3.27; F, 13.70; N, 10.10; O, 8.66; S, 5.78 | |
| CAS # | 1228591-30-7 | |
| Related CAS # |
|
|
| PubChem CID | 46209401 | |
| Appearance | White solid powder | |
| Density | 1.5±0.1 g/cm3 | |
| Index of Refraction | 1.660 | |
| LogP | 5.3 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 10 | |
| Rotatable Bond Count | 7 | |
| Heavy Atom Count | 39 | |
| Complexity | 957 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | O=C(NC1=CC(OC2=CC=C3N=C(NC(C4CC4)=O)SC3=C2C#N)=CC=C1F)CC5=CC=CC(C(F)(F)F)=C5 |
|
| InChi Key | OJFKUJDRGJSAQB-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C27H18F4N4O3S/c28-19-7-6-17(12-21(19)33-23(36)11-14-2-1-3-16(10-14)27(29,30)31)38-22-9-8-20-24(18(22)13-32)39-26(34-20)35-25(37)15-4-5-15/h1-3,6-10,12,15H,4-5,11H2,(H,33,36)(H,34,35,37) | |
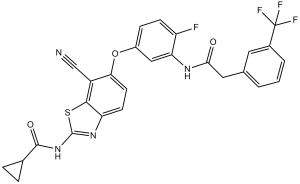
| Chemical Name | N-[7-cyano-6-[4-fluoro-3-[[2-[3-(trifluoromethyl)phenyl]acetyl]amino]phenoxy]-1,3-benzothiazol-2-yl]cyclopropanecarboxamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
c-Raf (IC50 = 1.4 nM); Braf (IC50 = 8.3 nM); Aurora B (IC50 = 66 nM); PDGFRβ (IC50 = 120 nM); PDGFRα (IC50 = 610 nM); FGFR3 (IC50 = 280 nM); TIE2 (IC50 = 740 nM); IKKβ (IC50 = 3700 nM); CDK1 (IC50 = 790 nM); CDK2 (IC50 = 580 nM); p38α (IC50 = 600 nM); GSK3β (IC50 = 500 nM); MEK1 (IC50 = 3700 nM) Pan-RAF kinases, including c-Raf (IC50: 3.2 nM), A-Raf (IC50: 4.4 nM), B-Raf wild-type (B-Raf WT, IC50: 5.0 nM), and B-Raf V600E mutant (IC50: 2.6 nM) [1] - Pan-RAF kinases, including B-Raf V600E (IC50: 2.8 nM), c-Raf (IC50: 3.5 nM), and A-Raf (IC50: 4.1 nM); no significant inhibition on other kinases (e.g., EGFR, VEGFR2) with IC50 > 1000 nM [2] |
| ln Vitro |
TAK-632 has an IC50 range of 120-790 nM and inhibits PDGFRβ, FGFR3, GSK3β, CDK2, P38α, PDGFRα, TIE2, and CDK1. The IC50 range for CHK1, IKKβ, and MEK1 is between 140 and 1700 nM. TAK-632 inhibits BRAF and CRAF after 1 hour of preincubation in an ATP competitive manner (at low ATP concentrations, BRAF IC50: 15 nM; CRAF: 8.1 nM). At high ATP concentrations, TAK-632 loses its biochemical activity against BRAF and CRAF, respectively, to IC50 values of 58 nM and 62 nM. In HMVII cells, TAK-632 exhibits potent inhibition of pMEK and pERK with IC50 values of 49 nM and 50 nM, respectively[1]. TAK-632 exhibits potent antiproliferative properties in both A375 and SK-MEL-2 cells (GI50 in A375 cells is 40–190 nM and GI50 in SK-MEL-2 cells is 190–250 nM)[2]. Against human cancer cell lines with RA F pathway activation: TAK-632 exhibited antiproliferative activity with IC50 values of 18 nM in A375 (B-Raf V600E melanoma), 25 nM in HT-29 (B-Raf V600E colorectal), 32 nM in SK-MEL-28 (B-Raf V600E melanoma), and 45 nM in HCT116 (K-Ras mutant colorectal) cells (evaluated by MTT assay). Western blot analysis showed that treatment with 50 nM TAK-632 for 4 hours reduced phosphorylated ERK (p-ERK) levels by ~80% in A375 cells and ~75% in HT-29 cells compared to vehicle control [1] - Against BRAF inhibitor-resistant melanoma cells: TAK-632 inhibited proliferation of A375-R (resistant to vemurafenib) and SK-MEL-28-R (resistant to dabrafenib) cells with IC50 values of 22 nM and 27 nM, respectively (CCK-8 assay). In A375-R cells, 30 nM TAK-632 treatment for 6 hours suppressed p-ERK levels by ~90% (Western blot) and reduced the expression of cyclin D1 (a downstream MAPK target) by ~70% (qPCR). Additionally, 50 nM TAK-632 induced apoptosis in A375-R cells, with apoptotic rate increasing from ~5% (control) to ~35% (Annexin V/PI staining) [2] |
| ln Vivo |
TAK-632 exhibits significantly increased oral absorption (at a dose of 25 mg/kg, AUC, 32.47 μg h/mL; F, 51.7%) and significantly improved solubility (740 μg/mL) in pH 6.8 phosphate buffer. The oral bioavailability of TAK-632 at 10 mg/kg in a dog PK study is also superior (F: 108%). Over a dose range of 1.9-24.1 mg/kg, oral single administration of TAK-632 inhibits pERK in tumors at 8 hours after its administration. Particularly, doses of 9.7-24.1 mg/kg of TAK-632 significantly reduce pERK levels to 11% of the control. Over a dose range of 3.9-24.1 mg/kg, TAK-632 demonstrates dose-dependent antitumor efficacy without significantly reducing body weight. Significant tumor regression is seen at doses of 9.7 mg/kg and 24.1 mg/kg (T/C=2.1% and 12.1%, respectively)[1]. In NRAS-mutant melanoma using a SK-MEL-2 xenograft model, TAK-632 exhibits potent antitumor efficacy when orally administered at 60 mg/kg once daily (T/C=37%, P<0.001) or at 120 mg/kg once daily (T/C=29%, P<0.001) for 21 days without severe toxicity[2]. In nude mouse xenograft models of A375 (B-Raf V600E melanoma): Oral administration of TAK-632 at 10 mg/kg, 30 mg/kg, and 60 mg/kg once daily for 21 days resulted in tumor growth inhibition (TGI) rates of 42%, 75%, and 91%, respectively. At 30 mg/kg, TAK-632 reduced p-ERK levels in tumor tissues by ~85% (immunohistochemistry, IHC) compared to vehicle control [1] - In nude mouse xenograft models of BRAF inhibitor-resistant melanoma: - A375-R model: Oral TAK-632 at 30 mg/kg and 60 mg/kg once daily for 28 days achieved TGI of 65% and 82%, respectively, while vemurafenib (60 mg/kg) only showed 12% TGI. IHC revealed that 30 mg/kg TAK-632 decreased p-ERK levels in tumor tissues by ~78% [2] - SK-MEL-28-R model: 60 mg/kg TAK-632 (oral, daily) for 24 days led to 78% TGI, with no significant weight loss (<5%) in mice [2] |
| Enzyme Assay |
Recombinant inactive MEK (K97R) is incubated with immunoprecipitated BRAF or CRAF at 30°C for 30 minutes in kinase reaction buffer containing ATP/Mg2+. TAK-632 is administered to RAS/RAF wild-type (A431, CsFb, and HeLa), KRAS-mutant (A549, HCT-116, and MIA PaCa-2), and NRAS-mutant (GAK, HMV-II, and SK-MEL-2) melanoma cells at the indicated concentrations for 2 hours. Western blot analysis is used to examine cell lysates[2]. RAF kinase activity assay (HTRF-based): The reaction system (25 μL total volume) contained recombinant human RAF kinase (c-Raf, A-Raf, B-Raf WT, or B-Raf V600E), 100 nM MEK1 (substrate), 1 μM ATP, and TAK-632 at concentrations ranging from 0.1 nM to 1000 nM. The mixture was incubated at 30°C for 60 minutes, then 25 μL of detection reagent (anti-phospho-MEK1 antibody and europium-labeled secondary antibody) was added. After incubation at room temperature for 30 minutes, fluorescence resonance energy transfer (FRET) signals were measured at excitation 340 nm and emission 620 nm/665 nm. The inhibition rate was calculated by comparing the signal ratio (620 nm/665 nm) of drug-treated groups to vehicle control, and IC50 values were derived from dose-response curves [1] - c-Raf/B-Raf V600E kinase activity assay: Recombinant c-Raf (5 ng/well) or B-Raf V600E (3 ng/well) was mixed with 50 μM ATP, 2 μg/mL peptide substrate (sequence corresponding to MEK1 phosphorylation site), and TAK-632 (0.05 nM–500 nM) in kinase buffer (25 mM Tris-HCl pH 7.5, 5 mM MgCl2, 1 mM DTT). The reaction was conducted at 37°C for 45 minutes, then terminated by adding 50 μL of 0.5 M HCl. Phosphorylated peptide was detected using a colorimetric antibody-based kit, and absorbance at 450 nm was measured. IC50 was calculated via nonlinear regression analysis [2] |
| Cell Assay |
The Sulforhodamine B assay or the CellTiter-Glo luminescent cell viability assay are both used to measure the viability of cells in three replicates. PCP software was used to determine the TAK-632 concentrations that resulted in 50% growth inhibition (GI50). Software called CalcuSyn is used to determine the combination index (CI). We carried out proliferation assays in various cell lines containing mutated BRAF, NRAS, or KRAS in order to look into the antiproliferative activity of TAK-632. TAK-632 and TAK-733 are cotreated for 72 hours with HMV-II, SK-MEL-2, or A375 cells. Measurements are made of cell viability. Calculations are made to determine the CI value at EC50. TAK-632 and TAK-733 are cotreated with A375 cells for 72 hours if they are transiently expressing NRASQ61K or ΔN-BRAF. Measurements are made of cell viability. Calculations are made to determine the CI value at EC50[2]. Antiproliferative assay (MTT method): - Cells (A375, HT-29, SK-MEL-28) were seeded into 96-well plates at 3×103 cells/well and cultured in complete medium (DMEM + 10% FBS) at 37°C, 5% CO2 for 24 hours. TAK-632 (0.1 nM–1000 nM, 10 concentrations) was added, and cells were incubated for another 72 hours. 20 μL MTT (5 mg/mL) was added to each well, followed by 4 hours of incubation. After removing supernatant, 150 μL DMSO was added to dissolve formazan crystals, and absorbance at 570 nm was measured. IC50 was calculated using GraphPad Prism software [1] - p-ERK Western blot assay: - A375 cells were seeded into 6-well plates at 2×105 cells/well and cultured for 24 hours. Cells were treated with TAK-632 (1 nM–100 nM) for 4 hours, then lysed with RIPA buffer (containing protease/phosphatase inhibitors). Protein concentration was determined by BCA assay, and 30 μg protein per lane was subjected to SDS-PAGE. Proteins were transferred to PVDF membranes, blocked with 5% non-fat milk for 1 hour, then incubated with primary antibodies against p-ERK (1:1000) and total ERK (1:2000) at 4°C overnight. After washing, membranes were incubated with HRP-conjugated secondary antibody (1:5000) for 1 hour, and signals were detected by ECL. Band intensity was quantified using ImageJ software [1] - BRAF inhibitor-resistant cell proliferation assay (CCK-8 method): - A375-R/SK-MEL-28-R cells were seeded into 96-well plates at 4×103 cells/well and synchronized with serum-free medium for 12 hours. TAK-632 (0.5 nM–500 nM) was added, and cells were cultured for 72 hours. 10 μL CCK-8 reagent was added, and absorbance at 450 nm was measured after 2 hours. IC50 values were calculated [2] - Apoptosis assay (Annexin V/PI staining): - A375-R cells were seeded into 6-well plates at 3×105 cells/well and treated with 50 nM TAK-632 for 48 hours. Cells were harvested, washed with PBS, and stained with Annexin V-FITC and PI according to the kit protocol. Apoptotic cells were analyzed by flow cytometry, and the percentage of Annexin V-positive/PI-negative (early apoptosis) and Annexin V-positive/PI-positive (late apoptosis) cells was calculated [2] |
| Animal Protocol |
Mice: The xenograft-implanted nude mice are employed. TAK-632 or vehicle are administered once daily for 21 straight days to mice with SK-MEL-2 xenografts (10 mice in each treatment group). Day 0 marks the start of the course of treatment. Two times a week, tumors are measured. Treatment options include vehicle, TAK-632 at 60 mg/kg (60 mpk), or TAK-632 at 120 mg/kg (120 mpk) once daily (QD) for three days in mice bearing SK-MEL-2 xenografts. After the final treatment, tumor xenografts are collected at the appropriate times and subjected to Western blot analysis. A single electrophoresis gel is used to combine various blots with dividing lines. The bars show the densitometric analysis of phospho-ERK normalized to the vehicle-treated control (mean±SD). Nude mouse A375 xenograft model: - Female BALB/c nude mice (6–7 weeks old, 18–22 g) were subcutaneously injected with 5×106 A375 cells (suspended in 100 μL PBS + 100 μL Matrigel) into the right flank. When tumors reached ~100 mm³, mice were randomly divided into 4 groups (n=6/group): vehicle control (0.5% methylcellulose + 0.1% Tween 80), TAK-632 10 mg/kg, 30 mg/kg, and 60 mg/kg. TAK-632 was dissolved in the vehicle, administered orally once daily for 21 days. Tumor volume (V = 0.5 × length × width²) and body weight were measured every 3 days. At the end of the experiment, tumors were excised for Western blot (p-ERK detection) [1] - Nude mouse BRAF inhibitor-resistant xenograft models: - A375-R model: Female nude mice (6–7 weeks old) were subcutaneously injected with 6×106 A375-R cells (100 μL PBS + 100 μL Matrigel) into the right flank. When tumors reached ~120 mm³, mice were grouped (n=6/group): vehicle, TAK-632 30 mg/kg, TAK-632 60 mg/kg, and vemurafenib 60 mg/kg. All drugs were administered orally once daily for 28 days. Tumor volume and body weight were measured every 2 days; tumors were collected for IHC (p-ERK staining) [2] - SK-MEL-28-R model: Mice were injected with 5×106 SK-MEL-28-R cells, and treated with TAK-632 60 mg/kg (oral, daily) for 24 days when tumors reached ~100 mm³. Tumor volume was measured every 3 days; no significant body weight loss was monitored [2] |
| ADME/Pharmacokinetics |
In SD rats (n=3/sex/dose): - Oral administration of TAK-632 (10 mg/kg): Peak plasma concentration (Cmax) = 125 ng/mL, time to Cmax (Tmax) = 1.5 hours, half-life (t1/2) = 4.2 hours, oral bioavailability (F) = 48%, clearance (CL) = 18 mL/min/kg, volume of distribution (Vd) = 5.8 L/kg [1] - Intravenous administration of TAK-632 (2 mg/kg): Cmax = 89 ng/mL, t1/2 = 3.8 hours, CL = 19 mL/min/kg [1] - In CD-1 mice (n=3/sex/dose): Oral TAK-632 (10 mg/kg) showed Cmax = 98 ng/mL, Tmax = 1 hour, t1/2 = 3.5 hours, F = 52% [1] |
| Toxicity/Toxicokinetics |
Acute toxicity in CD-1 mice: Oral administration of TAK-632 at doses up to 200 mg/kg (single dose) showed no mortality or significant toxicity (e.g., body weight loss <10%, normal behavior). Histopathological examination of liver and kidney revealed no abnormal lesions [1] - Subacute toxicity in SD rats: Oral TAK-632 (30 mg/kg, 60 mg/kg) once daily for 28 days: No significant changes in body weight, food intake, or hematological parameters (e.g., WBC, RBC, platelets). Serum biochemistry (ALT, AST, creatinine) was within normal range; no organ toxicity was observed via histology [1] - Plasma protein binding: In human plasma, TAK-632 showed a binding rate of 92% (equilibrium dialysis method); in rat and mouse plasma, binding rates were 90% and 88%, respectively [1] |
| References |
[1]. Discovery of a selective kinase inhibitor (TAK-632) targeting pan-RAF inhibition: design, synthesis, and biological evaluation of C-7-substituted 1,3-benzothiazole derivatives. J Med Chem. 2013 Aug 22;56(16):6478-94. [2]. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 2013 Oct 11. |
| Additional Infomation |
TAK-632 is a member of the class of benzothiazoles that is 1,3-benzothiazole substituted by (cyclopropanecarbonyl)amino, 4-fluoro-3-{2-[3-(trifluoromethyl)phenyl]acetamido}phenoxy, and cyano groups at positions 2, 6 and 7, respectively. It is a potent pan-RAF inhibitor with IC50 of 1.4, 2.4 and 8.3 nM for CRAF, BRAF(V600E), BRAF(WT), respectively. It has a role as a necroptosis inhibitor, a B-Raf inhibitor, an EC 2.7.11.26 (tau-protein kinase) inhibitor, an antineoplastic agent and an apoptosis inducer. It is a member of monofluorobenzenes, a member of benzothiazoles, an aromatic ether, a secondary carboxamide, a member of (trifluoromethyl)benzenes, a nitrile and a cyclopropylcarboxamide. TAK-632 is a rationally designed C-7-substituted 1,3-benzothiazole derivative targeting pan-RAF kinases. Its design aimed to overcome limitations of selective B-Raf inhibitors (e.g., acquired resistance due to c-Raf activation). Mechanistically, TAK-632 binds to the ATP-binding pocket of RAF kinases, inhibiting their catalytic activity and blocking the MAPK (Raf-MEK-ERK) signaling pathway, which is hyperactivated in many cancers [1] - BRAF inhibitor resistance (e.g., to vemurafenib, dabrafenib) in melanoma is often associated with reactivation of the MAPK pathway via c-Raf upregulation or alternative mutations. TAK-632’s pan-RAF inhibition activity enables it to suppress pathway activation in resistant cells, making it a potential therapeutic agent for BRAF inhibitor-resistant melanoma. Preclinical data in resistant xenograft models support its efficacy in overcoming resistance [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: 2.5 mg/mL (4.51 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (4.51 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 3: 2% DMSO+98% PEG 300: 5mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8034 mL | 9.0168 mL | 18.0336 mL | |
| 5 mM | 0.3607 mL | 1.8034 mL | 3.6067 mL | |
| 10 mM | 0.1803 mL | 0.9017 mL | 1.8034 mL |