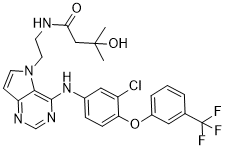
TAK-285 (TAK285), currently being investigated by Takeda company, is a dual HER2/EGFR(HER1) inhibitor with potential antitumor activity. Its IC50 values for HER2/1 inhibition are 17 nM and 23 nM. More than ten times more selective for HER1/2 than HER4, it is less effective against MEK1/5, Aurora B, Lck, c-Met, CSK, and other substances.
Physicochemical Properties
| Molecular Formula | C26H25CLF3N5O3 | |
| Molecular Weight | 547.96 | |
| Exact Mass | 547.159 | |
| Elemental Analysis | C, 56.99; H, 4.60; Cl, 6.47; F, 10.40; N, 12.78; O, 8.76 | |
| CAS # | 871026-44-7 | |
| Related CAS # |
|
|
| PubChem CID | 11620908 | |
| Appearance | White to off-white solid powder | |
| Density | 1.4±0.1 g/cm3 | |
| Boiling Point | 705.5±60.0 °C at 760 mmHg | |
| Melting Point | 167-169℃ | |
| Flash Point | 380.4±32.9 °C | |
| Vapour Pressure | 0.0±2.4 mmHg at 25°C | |
| Index of Refraction | 1.608 | |
| LogP | 3.56 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 9 | |
| Rotatable Bond Count | 9 | |
| Heavy Atom Count | 38 | |
| Complexity | 792 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | O=C(CC(C)(C)O)NCCN1C2C(=NC=NC=2NC2C=C(Cl)C(OC3C=C(C(F)(F)F)C=CC=3)=CC=2)C=C1 |
|
| InChi Key | ZYQXEVJIFYIBHZ-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C26H25ClF3N5O3/c1-25(2,37)14-22(36)31-9-11-35-10-8-20-23(35)24(33-15-32-20)34-17-6-7-21(19(27)13-17)38-18-5-3-4-16(12-18)26(28,29)30/h3-8,10,12-13,15,37H,9,11,14H2,1-2H3,(H,31,36)(H,32,33,34) | |
| Chemical Name | N-[2-[4-[3-chloro-4-[3-(trifluoromethyl)phenoxy]anilino]pyrrolo[3,2-d]pyrimidin-5-yl]ethyl]-3-hydroxy-3-methylbutanamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
HER2 (IC50 = 17 nM); EGFR/HER1 (IC50 = 23 nM); HER4 (IC50 = 260 nM); MEK1 (IC50 = 1.1 μM); Aurora B (IC50 = 1.7 μM) TAK-285 potently inhibits EGFR tyrosine kinase, including wild-type (IC₅₀ = 1.3 nM), EGFR L858R (IC₅₀ = 0.8 nM), and EGFR T790M (IC₅₀ = 2.9 nM) mutants [1] TAK-285 also inhibits HER2 (IC₅₀ = 4.2 nM) and HER4 (IC₅₀ = 6.7 nM) tyrosine kinases, with no significant effect on VEGFR2 or PDGFRβ (IC₅₀ > 1000 nM) [2] |
| ln Vitro |
Among the 34 kinases tested, TAK-285 only significantly inhibits HER4 with IC50 of 260 nM, slightly inhibits MEK1, MEK5, c-Met, Aurora B, Lck, CSK, and Lyn B with IC50 of 1.1 μM, 5.7 μM, 4.2 μM, 1.7 μM, 2.4 μM, 4.7 μM, and 5.2 μM, respectively, and displays no activity against other kinases with IC50 of >10 μM. TAK-285 shows significant growth inhibitory activity against BT-474 cells (HER2-overexpressing human breast cancer cell line) with GI50 of 17 nM.[1] TAK-285 exhibits comparable in vitro potency against EGFR and HER2 in comparison to SYR127063, a potent HER2 inhibitor. The inhibitory activity (IC50) of TAK-285 is not significantly affected by the mutations and shortened boundaries used for HER2-KD and EGFR-KD structure determination when compared to the full cytoplasmic domains of the wild-type proteins. TAK-285 exhibits a comparable binding mode to lapatinib in the active site when it comes to binding to the inactive conformation of EGFR.[2] TAK-285 dose-dependently inhibited the proliferation of EGFR/HER2-overexpressing tumor cell lines, including A431 (EGFR-overexpressing, IC₅₀ = 0.02 μM), NCI-H1975 (EGFR L858R/T790M, IC₅₀ = 0.05 μM), and SK-BR-3 (HER2-overexpressing, IC₅₀ = 0.08 μM). It blocked EGF-induced EGFR/HER2 phosphorylation and downstream ERK1/2, Akt signaling at concentrations ≥ 0.1 μM [1] TAK-285 induced apoptosis in NCI-H1975 cells with an EC₅₀ of 0.12 μM, upregulating cleaved caspase-3, -7, and PARP expression. It also suppressed clonogenicity of gefitinib-resistant NSCLC cells (PC-9/GR) with an IC₅₀ = 0.06 μM [2] In glioblastoma cells (U87MG) overexpressing EGFR, TAK-285 (0.1 μM) inhibited cell migration by ~65% and downregulated MMP-2 mRNA expression by ~58% [3] |
| ln Vivo |
TAK-285 has an oral bioavailability of 72.2% in mice and 97.7% in rats at a dose of 50 mg/kg. In the HER2-overexpressing BT-474 tumor xenograft mouse model, oral TAK-285 at 100 mg/kg twice daily for 14 days significantly reduces tumor growth without changing body weight, and the tumor/control (T/C) ratio is 29%. TAK-285, like the BT-474 model, shows dose-dependent inhibition of tumor growth of 4-1ST (HER2-overexpressing human gastric cancer tumor) xenografts in mice, with T/C of 44% and 11% at doses of 50 mg/kg and 100 mg/kg, twice daily, respectively, without causing a significant reduction in the mice's body weight. In addition, TAK-285 treatment results in tumor regression with T/C of -12% and -16% at doses of 25 mg/kg and 50 mg/kg, respectively, and dose-dependent growth inhibition of 4-1ST tumors in rats with T/C of 38% and 14% at doses of 6.25 mg/kg and 12.5 mg/kg. [1] Following oral administration of TAK-285, rats' brains contain a sizable amount of the drug in an unbound, pharmacologically active form (roughly 20% of its free plasma level), suggesting that TAK-285 may be used to treat CNS cancers and their metastases.[3] TAK-285 significantly inhibited tumor growth in nude mice bearing NCI-H1975 xenografts when administered orally at 20 mg/kg/day for 21 days. Tumor volume was reduced by ~80% compared to the control group, and intratumoral EGFR phosphorylation was nearly completely blocked [1] TAK-285 prolonged median survival of nude mice bearing SK-BR-3 xenografts by 50% when given orally at 25 mg/kg/day for 28 days. It also downregulated Ki-67 (proliferation marker) expression in tumor tissues by ~72% [2] In a mouse model of glioblastoma xenografts (U87MG), TAK-285 (30 mg/kg/day, oral) reduced tumor volume by ~68% and inhibited intracranial tumor progression [3] |
| Enzyme Assay |
The cytoplasmic domains of human HER2 (amino acids 676–1255) and human EGFR (amino acids 669–1210) are expressed via a baculovirus expression system as N-terminal peptide (DYKDDDD)-tagged protein. Anti-FLAG M2 affinity gel is used to separate the expressed EGFR and HER2 kinases. Radiolabeled [γ- 32 P]ATP is used in 96-well plates for the EGFR and HER2 kinase assays. Each purified cytoplasmic domain (0.25 μg/mL EGFR or HER2) in a total volume of 50 μL, 50 μM ATP, 5 ug/mL poly(Glu)-Tyr (4:1), and 0.9 uCi of [γ- 32 P]ATP per reaction are added to the 50 μL of Tris-HCl (pH 7.5), 0.01% Tween 20, and 2 mM DTT. The enzyme is incubated with increasing concentrations of TAK-285 for five minutes at room temperature before the reaction is measured to determine the IC50 value for enzyme inhibition. ATP is added to start the kinase reactions. The reactions are halted by adding 10% (final concentration) trichloroacetic acid after 10 minutes at room temperature. After using a cell harvester to filter the γ- 32 P phosphorylated proteins in a harvest plate, 3% phosphoric acid is used to remove any remaining [γ- 32 P]ATP. MicroScint0 (25 μL) is added to the plates after they have dried. A TopCount scintillation counter measures radioactivity. The nonlinear regression analysis of the percent inhibitions is used to compute the IC50 values. Recombinant EGFR (wild-type, L858R, T790M) and HER2/HER4 kinase domains were individually incubated with ATP and specific peptide substrates in the presence of serial dilutions of TAK-285. Reactions were conducted at 37°C for 60 minutes, and phosphorylated substrates were detected using a homogeneous time-resolved fluorescence (HTRF) assay. Inhibition rates were calculated by comparing fluorescence intensity with vehicle controls, and IC₅₀ values were derived from dose-response curves [1] To assess selectivity, recombinant VEGFR2 and PDGFRβ kinase domains were tested using the same protocol. Reaction conditions were identical, and IC₅₀ values were determined to confirm preferential targeting of EGFR family kinases [2] |
| Cell Assay |
For five days, the cells are continuously treated with TAK-285 at different concentrations. A particle analyzer is used to count the number of live cells. A431, NCI-H1975, and SK-BR-3 cells were seeded in 96-well plates at 5×10³ cells/well and treated with TAK-285 (0.001-0.5 μM) for 72 hours. Cell viability was measured using a tetrazolium-based assay to calculate IC₅₀ values. For Western blot analysis, cells were treated with 0.05-0.2 μM drug and stimulated with EGF, then lysed and probed with antibodies against phosphorylated EGFR/HER2, ERK1/2, Akt, and GAPDH [1] NCI-H1975 and PC-9/GR cells were treated with TAK-285 (0.05-0.2 μM) for 48 hours. Apoptosis was detected by Annexin V-FITC/PI staining, and cleaved caspase-3/-7/PARP expression was analyzed by Western blot. Clonogenic assays were performed by treating cells with 0.03-0.1 μM drug for 14 days, followed by fixation, staining, and colony counting [2] U87MG cells were treated with TAK-285 (0.05-0.2 μM) for 24 hours. Migration assays were performed using Boyden chambers, and MMP-2 mRNA expression was quantified by RT-PCR [3] |
| Animal Protocol |
Female BALB/c nu/nu mice bearing BT-474 or 4-1ST xenografts, and female nude rats (F344/N Jcl-rnu) bearing 4-1ST xenografts ~100 mg/kg/day Orally twice daily Nude mice bearing NCI-H1975 xenografts (100-150 mm³) were randomly divided into control and treatment groups. TAK-285 was suspended in 0.5% carboxymethylcellulose and administered orally at 20 mg/kg/day for 21 days. Tumor volume was measured every 3 days, and mice were euthanized to collect tumors for Western blot analysis of EGFR phosphorylation [1] Nude mice bearing SK-BR-3 xenografts were treated with TAK-285 orally at 25 mg/kg/day for 28 days. Survival time was recorded daily, and tumor tissues were processed for immunohistochemical staining of Ki-67 [2] Nude mice were implanted with U87MG glioblastoma cells intracranially. Seven days later, mice were treated with TAK-285 orally at 30 mg/kg/day for 24 days. Mice were euthanized, and brains were harvested to measure tumor volume and analyze MMP-2 expression by immunohistochemistry [3] |
| ADME/Pharmacokinetics |
TAK-285 had an oral bioavailability of ~70% in mice after a single dose of 20 mg/kg. The plasma half-life was approximately 8.2 hours, and the maximum plasma concentration (Cmax) was 3.8 μg/mL achieved at 1.5 hours post-administration [1] In rats, oral administration of TAK-285 at 25 mg/kg resulted in an AUC₀-24h of 45.6 μg·h/mL. The drug penetrated the blood-brain barrier effectively, with a brain-to-plasma concentration ratio of ~0.7 [3] |
| Toxicity/Toxicokinetics |
Mice treated with TAK-285 at 20 mg/kg/day for 21 days showed mild weight loss (~6%) but no significant liver or kidney toxicity. Serum ALT, AST, and creatinine levels were within normal ranges [1] The plasma protein binding rate of TAK-285 was ~95% in human plasma as determined by equilibrium dialysis. In long-term toxicity studies (28 days, 25 mg/kg/day, oral), rats showed no severe hematological or gastrointestinal toxicities [2] |
| References |
[1]. J Med Chem . 2011 Dec 8;54(23):8030-50. [2]. J Biol Chem . 2011 May 27;286(21):18756-65. [3]. Brain Res Bull . 2012 Mar 10;87(4-5):413-9. |
| Additional Infomation |
EGFR/HER2 Kinase Inhibitor TAK-285 is an orally bioavailable, small molecule and dual kinase inhibitor of human epidermal growth factor receptors 1 (EGFR/ErbB1) and 2 (HER2/ErbB2), with potential antineoplastic activity. EGFR/HER2 kinase inhibitor TAK-285 binds to and inhibits EGFR and HER2, which may result in the inhibition of tumor growth and angiogenesis, and tumor regression in EGFR/HER2-expressing tumors. This agent may be active against EGFR/HER2-expressing tumor cells that are resistant to trastuzumab. EGFR and HER2, receptor tyrosine kinases overexpressed in a variety of tumor cell types, play major roles in tumor cell proliferation and tumor vascularization. In addition, TAK-285 appears to pass the blood brain barrier (BBB) and does not appear to be a substrate for efflux pumps. TAK-285 is an irreversible pan-HER tyrosine kinase inhibitor that covalently binds to the ATP-binding site of EGFR, HER2, and HER4, blocking downstream signaling pathways involved in tumor proliferation, survival, and migration [1] It was developed to overcome T790M-mediated resistance to first-generation EGFR inhibitors in NSCLC and shows potential for treating EGFR-mutant glioblastoma due to its blood-brain barrier penetration ability [3] TAK-285 exhibits synergistic antitumor effects when combined with chemotherapy agents (e.g., paclitaxel) in preclinical models, supporting its potential for combination therapy [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (4.56 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (4.56 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (4.56 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8250 mL | 9.1248 mL | 18.2495 mL | |
| 5 mM | 0.3650 mL | 1.8250 mL | 3.6499 mL | |
| 10 mM | 0.1825 mL | 0.9125 mL | 1.8250 mL |