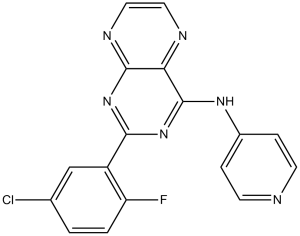
SD-208 (SD208; SD 208; TGF-β RI Kinase Inhibitor V) is an orally bioavailable and selective TGF-βRI (ALK5) inhibitor with potential antitumor activity. It inhibits ALK5 with an IC50 of 48 nM, and displayed >100-fold selectivity for TGF-βRI over TGF-βRII. It shows potent anti-proliferative activity in vitro against various cancer cell lines such as murine and human glioma cells.
Physicochemical Properties
| Molecular Formula | C17H10CLFN6 | |
| Molecular Weight | 352.75 | |
| Exact Mass | 352.063 | |
| Elemental Analysis | C, 57.88; H, 2.86; Cl, 10.05; F, 5.39; N, 23.82 | |
| CAS # | 627536-09-8 | |
| Related CAS # |
|
|
| PubChem CID | 10316032 | |
| Appearance | Light yellow to yellow solid powder | |
| Density | 1.5±0.1 g/cm3 | |
| Boiling Point | 460.4±45.0 °C at 760 mmHg | |
| Flash Point | 232.2±28.7 °C | |
| Vapour Pressure | 0.0±1.1 mmHg at 25°C | |
| Index of Refraction | 1.717 | |
| LogP | 2.69 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 7 | |
| Rotatable Bond Count | 3 | |
| Heavy Atom Count | 25 | |
| Complexity | 437 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | ClC1C([H])=C([H])C(=C(C=1[H])C1=NC2C(C(=N1)N([H])C1C([H])=C([H])N=C([H])C=1[H])=NC([H])=C([H])N=2)F |
|
| InChi Key | BERLXWPRSBJFHO-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C17H10ClFN6/c18-10-1-2-13(19)12(9-10)15-24-16-14(21-7-8-22-16)17(25-15)23-11-3-5-20-6-4-11/h1-9H,(H,20,22,23,24,25) | |
| Chemical Name | 2-(5-chloro-2-fluorophenyl)-N-4-pyridinyl-4-pteridinamine | |
| Synonyms | SD-208; SD 208; 2-(5-chloro-2-fluorophenyl)-N-(pyridin-4-yl)pteridin-4-amine; SD 208; SD208; 2-(5-chloro-2-fluorophenyl)-N-pyridin-4-ylpteridin-4-amine; CHEMBL238125; MFCD11519969; TGF-β RI Kinase Inhibitor V; SD208; | |
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
TGF-βRI (ALK5) (IC50 = 48 nM) SD-208 specifically targets transforming growth factor-beta type I receptor (TGF-β RI/ALK5) (ALK5 IC50 = 170 nM) [1] SD-208 shows no significant inhibition of other kinases (PKA, PKC, ERK1/2: IC50 > 10 μM) [1] |
| ln Vitro |
In both human LN-308 and murine SMA-560 glioma cells, SD-208 increases immunogenicity while blocking constitutive, TGF-beta-evoked, and cell growth[1]. In vitro, SD-208 promotes migration, invasiveness, and epithelial-to-mesenchymal transdifferentiation through blocking TGF-beta-induced phosphorylation of Smad2 and Smad3[2]. Additionally, TGF-β's stimulating influence on the migration and proliferation of neointimal smooth muscle-like cells (SMLCs) in vitro is eliminated by SD-208[3]. In human (U87MG) and murine (C6) glioma cells, SD-208 (5 μM) inhibits cell proliferation by 55-60% after 72 hours (CCK-8 assay). It reduces cell invasion by 70% (Matrigel Transwell assay) and downregulates TGF-β target genes (PAI-1, CTGF) by 65% and 62% at mRNA level. It also enhances immunogenicity by increasing MHC class I molecule expression (2.2-fold) on U87MG cells [1] - In murine (4T1) and human (MDA-MB-231) breast cancer cells, SD-208 (10 μM) inhibits cell proliferation by 63-68% after 48 hours (MTT assay) and reduces cell migration by 65% (wound-healing assay). It induces apoptosis, with Annexin V-positive cells increasing from 8% to 36% (4T1 cells) after 72 hours, and downregulates mesenchymal marker vimentin by 70% at protein level [2] - In murine aortic smooth muscle cells (MASMCs), SD-208 (2 μM) inhibits TGF-β1-induced cell proliferation by 58% after 48 hours and migration by 62% after 24 hours (Boyden chamber assay). It downregulates α-SMA expression (fibrosis marker) by 65% and collagen type I synthesis by 60% [3] - In normal human astrocytes and mammary epithelial cells, SD-208 shows low toxicity at concentrations up to 25 μM (cell viability > 85% vs. control) [1][2] |
| ln Vivo |
Mice bearing SMA-560 gliomas have a considerably longer median survival when treated with SD-208 (1 mg/mL, po)[1]. SD-208 (60 mg/kg/d, po) prevents the formation of primary R3T tumors in syngeneic 129S1 mice and decreases the quantity and size of lung metastases[2]. SD-208 successfully inhibits the development of intimal hyperplasia of transplant arteriosclerosis (TA) in the mouse aortic allograft model[3]. Treatment of syngeneic R3T or 4T1 tumor-bearing mice with orally given SD-208 inhibited primary tumor growth as well as the number and size of metastases. In contrast, SD-208 failed to inhibit R3T tumor growth or metastasis in athymic nude mice. [2] The intimal hyperplasia of the SD-208-treated group was significantly reduced compared with the vehicle-treated control group (32% and 48% reduction for 40 mg/kg and 60 mg/kg SD-208 compared with the controls, respectively [n = 5], p < 0.05). SD-208 reduced SMLC proliferation and the production of intimal collagen by 21% and 75%, respectively, in the grafts. SD-208 also abolished the promoting effect of TGF-β on SMLC proliferation and migration but did not affect TGF-β inhibition of VSMCs in vitro. CTGF, a protein downstream of TGF-β, was downregulated with the inhibition of Smad3 phosphorylation by SD-208, both in vitro and in vivo. Moreover, we found that the endogenous Smad3 in SMLCs was upregulated from 2 weeks after transplantation and was 64% higher than in VSMCs at 8 weeks. Conclusion: These results demonstrate that SD-208 can effectively reduce the formation of intimal hyperplasia of TA in the murine aortic allograft model.[3] In nude mice bearing subcutaneous U87MG glioma xenografts, oral administration of SD-208 (100 mg/kg/day for 28 days) significantly inhibits tumor growth. Tumor volume was reduced by 62% compared to vehicle controls, and median survival was prolonged from 32 days to 54 days. Tumor tissues show downregulated p-Smad2 (75% reduction) and Ki-67 (55% reduction) [1] - In BALB/c mice with orthotopic 4T1 breast cancer, oral SD-208 (75 mg/kg/day for 21 days) inhibits primary tumor growth (volume reduced by 58%) and reduces lung metastatic nodules by 70% compared to vehicle. It suppresses TGF-β-mediated EMT in metastatic tissues (vimentin downregulated by 68%) [2] - In C57BL/6 mice with aortic allografts, oral SD-208 (50 mg/kg/day for 30 days) inhibits intimal hyperplasia. Intimal thickness was reduced by 63% compared to vehicle, and graft tissues show decreased α-SMA-positive cells (60% reduction) and collagen deposition (55% reduction) [3] |
| Enzyme Assay |
Transforming Growth Factor β Reporter Assays.[1] Intracellular TGF-β signaling was assessed by reporter assays using pGL2 3TP-Luc or pGL3 SBE-2 Luc (17) reporter gene plasmids. The pGL2 3TP-Luc construct contains a synthetic promoter composed of a TGF-β–responsive plasminogen activator inhibitor 1 promoter fragment inserted downstream of three phorbol ester-responsive elements. The pGL3 SBE-2-Luc reporter contains two copies of the Smad-binding element GTCTAGAC. LN-308 and SMA-560 cells were transfected using FuGene. At 24 hours after transfection, the cells were pretreated in serum-containing medium with SD-208 for 12 hours (1 μmol/L). TGF-β1 (5 ng/mL) was then added for another 16 hours. The cells were lysed and transferred to a LumiNunc plate, and luminescence was measured in a LumimatPlus, using a luciferase assay substrate. For T cell assays, 5 × 106 freshly isolated PBLs were cotransfected with 4.5 μg of pGL2–3TP-Luc or pGL3-SBE-2 Luc reporter gene plasmid and 0.5 μg of pRL-CMV, using the Nucleofector device and the cell type-specific human T-cell Nucleofector kit. IL-2 (50 units/mL) was added 4 hours after nucleofection, and the cells were pretreated with SD-208 for 1 hour before TGF-β1 (5 ng/mL) was added for another 16 hours. The respective activities of firefly and Renilla reniformis luciferase were determined sequentially using the Firelite dual luminescence reporter gene assay. Counts obtained from the measurement of firefly luciferase were normalized with respect to pRL-CMV. ALK5 kinase activity assay: Purified recombinant human ALK5 was incubated with Smad2-derived substrate peptide and SD-208 (0.1 nM-10 μM) in assay buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl₂, 1 mM DTT, 0.1 mM ATP) at 30°C for 60 minutes. Phosphorylated substrate was detected by radiolabeled ATP counting, and IC50 values were calculated from dose-response curves [1] - Kinase selectivity assay: SD-208 (10 μM) was screened against a panel of 30+ kinases (including PKA, PKC, ERK1/2, EGFR) using respective substrate peptides and assay buffers. Kinase activity was quantified by colorimetric assay, with no significant off-target inhibition (>50% activity reduction) observed [1] |
| Cell Assay |
Proliferation.[1] Glioma cells were cultured in the absence or presence of SD-208 (1 μmol/L) for 48 hours. The cells were pulsed for the last 24 hours with [methyl-3H]thymidine (0.5 μCi) and harvested, and incorporated radioactivity was determined in a liquid scintillation counter. Epithelial-to-mesenchymal transdifferentiation. [2] For assessment of subcellular F-actin fiber distribution, cells were washed with PBS and fixed with buffered formalin for 10 minutes. Following washes with PBS, cells were permeabilized using 0.1% (v/v) Triton X-100 in PBS for 5 minutes and then incubated with 0.165 μmol/L Alexa Fluor 488–conjugated phalloidin with 1% (w/v) bovine serum albumin in PBS for 20 minutes at 20°C in the dark. For E-cadherin immunostaining, cells were washed with PBS and fixed for 5 minutes using methanol precooled to −20°C. Air-dried slides were then incubated with 5% (v/v) goat serum for 20 minutes at room temperature followed by incubation with 2 μg/mL mouse monoclonal anti-E-cadherin antibody in 2.5% (v/v) goat serum for 1 hour at room temperature. The cells were then washed 3 × 5 minutes with PBS followed by incubation with 2 μg/mL rhodamine-conjugated goat anti-mouse IgG for 45 minutes in the dark. In both cases, stained dishes were then washed 3 × 5 minutes with PBS, mounted using Vectashield mounting medium, and viewed using a Zeiss epifluorescence microscope (model 090477) equipped with a MTI charge-coupled device camera.[2] Western blot analysis. [2] For detection of Smad proteins, semiconfluent cell cultures were lysed in situ using buffer composed of 150 mmol/L NaCl, 10 mmol/L Tris-HCl (pH 8.0), 1 mmol/L EDTA, 1 mmol/L EGTA, and 1% (v/v) Triton X-100 in the presence of protease inhibitors (Complete Mini Protease Inhibitor Cocktail Tablets) for 30 minutes at 4°C. Cell lysates were subjected to Western blot analysis as described previously (42). Activated Smad2 (pSmad2) and activated Smad3 (pSmad3) were detected using our own rabbit anti-pSmad2 and anti-pSmad3 antibodies at a 1:1,000 dilution. Total Smad2, Smad3, and Smad4 were detected using rabbit anti-Smad2, rabbit anti-Smad3, and mouse anti-Smad4 antibodies, respectively, at a 1:500 dilution.[2] In vitro cell migration and invasion assays. [2] For migration assays, uncoated polyethylene terephthalate track etched membrane (24-well insert; pore size, 8 μm) inserts were equilibrated by adding 0.5 mL cell culture medium to the upper and lower chambers followed by incubation at 37°C for 2 hours. For invasion assays, BD Biocoat Growth Factor Reduced Matrigel Invasion Chambers (24-well insert; pore size, 8 μm) were rehydrated by adding 0.5 mL warm (37°C) culture medium to the upper chambers followed by 2-hour incubation at 37°C. For both assays, medium used for equilibration was removed and 105 cells were plated in the upper chambers. TGF-β1 (100 pmol/L, 2.5 ng/mL), SD-093 (1 μmol/L), both agents, or vehicle only were added to both upper and lower chambers. Following a 24-hour incubation at 37°C, cells in suspension were aspirated, the inserts were washed twice with PBS, and the cells adherent to the top of the inserts were removed by scraping the upper surface of the membrane with a cotton tip applicator. The cells that had migrated to the underside of the inserts were fixed and stained using the DiffQuick staining kit. Cells in 10 random squares of 100 × 100 μm in each well were counted at ×200 magnification using quadruplicate wells per assay condition and the results were expressed as number of cells/mm2. Glioma cell proliferation/immunogenicity assay: U87MG and C6 cells were seeded in 96-well plates (proliferation) or 6-well plates (invasion/immunogenicity) at 3×10³ cells/well or 2×10⁵ cells/well respectively. Cells were treated with SD-208 (1-10 μM) for 48-72 hours. CCK-8 assay assessed proliferation; Matrigel Transwell assay evaluated invasion; qPCR analyzed PAI-1/CTGF mRNA levels; flow cytometry detected MHC class I expression [1] - Breast cancer cell proliferation/apoptosis assay: 4T1 and MDA-MB-231 cells were seeded in 96-well plates (proliferation) or 6-well plates (apoptosis/migration) at 3×10³ cells/well or 2×10⁵ cells/well respectively. Cells were treated with SD-208 (5-20 μM) for 48-72 hours. MTT assay measured proliferation; Annexin V-FITC/PI staining quantified apoptosis; wound-healing assay evaluated migration; Western blot detected vimentin [2] - Vascular smooth muscle cell assay: MASMCs were seeded in 96-well plates (proliferation) or Boyden chambers (migration) at 3×10³ cells/well or 5×10⁴ cells/chamber respectively. Cells were pretreated with SD-208 (0.5-5 μM) for 1 hour, then stimulated with TGF-β1 (10 ng/mL) for 24-48 hours. CCK-8 assay assessed proliferation; migrated cells were stained and counted; Western blot detected α-SMA; ELISA measured collagen type I synthesis [3] |
| Animal Protocol |
Dissolved in water; 1 mg/mL; p.o. VM/Dk mice bearing SMA-560 tumors Survival Studies In vivo.[1] VM/Dk cice of 6 to 12 weeks of age were used for the survival experiments. The experiments were performed according to the German animal protection law. Groups of eight mice were anesthesized before all intracranial procedures and placed in a stereotaxic fixation device. A burr hole was drilled in the skull 2 mm lateral to the bregma. The needle of a Hamilton syringe was introduced to a depth of 3 mm. SMA-560 cells [5 × 103 cells] resuspended in a volume of 2 μL of PBS were injected into the right striatum. Three days later, the mice were allowed to drink SD-208 at 1 mg/mL in deionized water. The mice were observed daily and, in the survival experiments, sacrificed on development of neurologic symptoms.[1] Animal experiments. [2] For in vivo studies, SD-208 was suspended in 1% (w/v) methylcellulose in water. Descriptive pharmacokinetic variables were determined by standard model-independent methods. Noncompartmental analysis was done using WinNonlin version 4.0.1. Because individual mice were not used to describe a full profile, variables were calculated using mean data. Samples with SD-208 concentration below quantifiable limits (10 ng/mL) were assigned the value of 0 for the analysis. Nominal time points were used for all calculations. AUC(0-8) is the area under the plasma concentration-time curve from time 0 to 8 hours for animals dosed with SD-208. BALB/c (H-2(d)) donor aortas were transplanted into C57BL/6 (H-2(b)) recipients, and the mice then received different doses (40 or 60 mg/kg) of SD-208 or control vehicle by daily gavage for 8 weeks. The grafts were analyzed by histology and morphometry at 1, 2, 4, 6 and 8 weeks after transplantation. The effects of TGF-β and SD-208 on neointimal smooth muscle-like cell (SMLC) and vascular smooth muscle cell (VSMC) proliferation and migration were evaluated, and the expression levels of Smad3, P-Smad3, connective tissue growth factor (CTGF) and collagen I were determined by in vitro experiments.[3] Nude mouse glioma xenograft model: 6-8 weeks old nude mice were subcutaneously inoculated with U87MG cells (5×10⁶ cells/mouse). When tumors reached ~100 mm³, mice were randomly divided into vehicle and SD-208 groups. The drug was suspended in 0.5% carboxymethylcellulose sodium and administered orally at 100 mg/kg/day for 28 days. Vehicle group received carboxymethylcellulose sodium. Tumor volume was measured every 3 days; median survival was recorded; tumor tissues were analyzed by Western blot (p-Smad2) and Ki-67 immunostaining [1] - BALB/c mouse breast cancer model: 6-8 weeks old BALB/c mice were orthotopically implanted with 4T1 cells (1×10⁶ cells/mouse) into mammary fat pads. One day post-implantation, SD-208 was suspended in 0.5% carboxymethylcellulose sodium and administered orally at 75 mg/kg/day for 21 days. Vehicle group received carboxymethylcellulose sodium. Primary tumor volume was measured weekly; lungs were collected to count metastatic nodules; Western blot detected vimentin in tumor tissues [2] - Mouse aortic allograft model: Aortic segments from C57BL/6 mice were transplanted into BALB/c mice. One day post-transplantation, SD-208 was suspended in 0.5% carboxymethylcellulose sodium and administered orally at 50 mg/kg/day for 30 days. Vehicle group received carboxymethylcellulose sodium. Grafts were harvested for histomorphometric analysis (intimal thickness); Masson’s trichrome staining evaluated collagen deposition; α-SMA immunostaining counted positive cells [3] |
| Toxicity/Toxicokinetics |
In vitro, SD-208 shows low toxicity to normal human cells (astrocytes IC50 > 25 μM; mammary epithelial cells IC50 > 30 μM) [1][2] - In in vivo studies, oral administration of SD-208 at tested doses (50-100 mg/kg/day) causes no significant body weight loss (<5% vs. baseline) or overt lethality in mice [1][2][3] - No significant changes in liver function (ALT, AST) or renal function (creatinine, BUN) were observed in SD-208-treated mice compared to vehicle controls [1][3] - Plasma protein binding rate of SD-208 is 89-92% in mice (in vitro plasma binding assay) [1] |
| References |
[1]. SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004 Nov 1;64(21):7954-61. [2]. Inhibition of growth and metastasis of mouse mammary carcinoma by selective inhibitor of transforming growth factor-beta type I receptor kinase in vivo. Clin Cancer Res. 2006 Jul 15;12(14 Pt 1):4315-30. [3]. Inhibition of intimal hyperplasia in murine aortic allografts by the oral administration of the transforming growth factor-beta receptor I kinase inhibitor SD-208. J Heart Lung Transplant. 2014 Jun;33(6):654-61. |
| Additional Infomation |
The cytokine transforming growth factor (TGF)-beta, by virtue of its immunosuppressive and promigratory properties, has become a major target for the experimental treatment of human malignant gliomas. Here we characterize the effects of a novel TGF-beta receptor (TGF-betaR) I kinase inhibitor, SD-208, on the growth and immunogenicity of murine SMA-560 and human LN-308 glioma cells in vitro and the growth of and immune response to intracranial SMA-560 gliomas in syngeneic VM/Dk mice in vivo. SD-208 inhibits the growth inhibition of TGF-beta-sensitive CCL64 cells mediated by recombinant TGF-beta1 or TGF-beta2 or of TGF-beta-containing glioma cell supernatant at an EC(50) of 0.1 mumol/L. SD-208 blocks autocrine and paracrine TGF-beta signaling in glioma cells as detected by the phosphorylation of Smad2 or TGF-beta reporter assays and strongly inhibits constitutive and TGF-beta-evoked migration and invasion, but not viability or proliferation. Peripheral blood lymphocytes or purified T cells, cocultured with TGF-beta-releasing LN-308 glioma cells in the presence of SD-208, exhibit enhanced lytic activity against LN-308 targets. The release of interferon gamma and tumor necrosis factor alpha by these immune effector cells is enhanced by SD-208, whereas the release of interleukin 10 is reduced. SD-208 restores the lytic activity of polyclonal natural killer cells against glioma cells in the presence of recombinant TGF-beta or of TGF-beta-containing glioma cell supernatant. The oral bioavailability of SD-208 was verified by demonstrating the inhibition of TGF-beta-induced Smad phosphorylation in spleen and brain. Systemic SD-208 treatment initiated 3 days after the implantation of SMA-560 cells into the brains of syngeneic VM/Dk mice prolongs their median survival from 18.6 to 25.1 days. Histologic analysis revealed no difference in blood vessel formation, proliferation, or apoptosis. However, animals responding to SD-208 showed an increased tumor infiltration by natural killer cells, CD8 T cells, and macrophages. These data define TGF-beta receptor I kinase inhibitors such as SD-208 as promising novel agents for the treatment of human malignant glioma and other conditions associated with pathological TGF-beta activity.[1] Purpose: Transforming growth factor-beta (TGF-beta) suppresses tumor development by inhibiting cellular proliferation, inducing differentiation and apoptosis, and maintaining genomic integrity. However, once tumor cells escape from the tumor-suppressive effects of TGF-beta, they often constitutively overexpress and activate TGF-beta, which may promote tumor progression by enhancing invasion, metastasis, and angiogenesis and by suppressing antitumor immunity. The purpose of this study was to test this hypothesis using TGF-beta pathway antagonists. Experimental design: We examined the effects of selective TGF-beta type I receptor kinase inhibitors, SD-093 and SD-208, on two murine mammary carcinoma cell lines (R3T and 4T1) in vitro and in vivo. Results: Both agents blocked TGF-beta-induced phosphorylation of the receptor-associated Smads, Smad2 and Smad3, in a dose-dependent manner, with IC50 between 20 and 80 nmol/L. TGF-beta failed to inhibit growth of these cell lines but stimulated epithelial-to-mesenchymal transdifferentiation, migration, and invasiveness into Matrigel in vitro. These effects were inhibited by SD-093, indicating that these processes are partly driven by TGF-beta. Treatment of syngeneic R3T or 4T1 tumor-bearing mice with orally given SD-208 inhibited primary tumor growth as well as the number and size of metastases. In contrast, SD-208 failed to inhibit R3T tumor growth or metastasis in athymic nude mice. Moreover, in vitro anti-4T1 cell cytotoxic T-cell responses of splenocytes from drug-treated animals were enhanced compared with cells from control animals. In addition, SD-208 treatment resulted in a decrease in tumor angiogenesis. Conclusion: TGF-beta type I receptor kinase inhibitors hold promise as novel therapeutic agents for metastatic breast cancer.[2] Background: Transforming growth factor-beta (TGF-β) plays a significant role in the pathogenesis of the intimal hyperplasia of transplant arteriosclerosis (TA). The aim of this study was to evaluate the efficacy of an oral inhibitor of TGF-β receptor I kinase (SD-208) on the development of TA.[3] SD-208 is a potent, selective small-molecule inhibitor of TGF-β type I receptor (ALK5) [1] - Its mechanism of action involves competitive binding to the ATP-binding pocket of ALK5, inhibiting its kinase activity and blocking downstream Smad2/3 phosphorylation, thereby suppressing TGF-β-mediated cell proliferation, migration, invasion, EMT, and fibrosis [1][2][3] - SD-208 exhibits in vitro anti-tumor (glioma, breast cancer) and anti-fibrotic (vascular smooth muscle cells) activities, as well as in vivo anti-tumor, anti-metastatic, and anti-transplant rejection effects [1][2][3] - It enhances the immunogenicity of glioma cells, supporting potential combination with immunotherapy [1] - SD-208 is widely used as a tool compound to study TGF-β/ALK5 signaling in cancer, organ transplantation, and fibroproliferative diseases [1][2][3] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: 0.91 mg/mL (2.58 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 9.1 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 0.91 mg/mL (2.58 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 9.1 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 0.91 mg/mL (2.58 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 9.1 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 1% methylcellulose:8 mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8349 mL | 14.1743 mL | 28.3487 mL | |
| 5 mM | 0.5670 mL | 2.8349 mL | 5.6697 mL | |
| 10 mM | 0.2835 mL | 1.4174 mL | 2.8349 mL |