SC66 is a novel, potent and allosteric inhibitor of AKT with potential anticancer activity, it reduces cell viability in a dose- and time-dependent manner, inhibits colony formation and induces apoptosis in hepatocellular carcinoma (HCC) cells. With IC50 values of 0.77, 2.85, and 0.47 μg/ml in HepG2, Huh7, and Hep3B cells, respectively, SC66 demonstrates a dual inhibitory function toward AKT activity. In hepatocellular carcinoma (HCC) cells, SC66 inhibits colony formation, decreases cell viability in a dose- and time-dependent manner, and triggers apoptosis. SC66 treatment led to a reduction in total and phospho-AKT levels. Reactive oxygen species (ROS) and DNA damage were both induced by SC66. Both the effects of doxorubicin, a traditional chemotherapeutic, and those of everolimus, a targeted agent, were markedly enhanced by SC66. Hep3B cell tumor growth was inhibited by SC66 in vivo in xenograft models; an analogous mechanism was seen in the in vitro model. HCC cells responded favorably to SC66's antitumor properties.
Physicochemical Properties
| Molecular Formula | C18H16N2O | |
| Molecular Weight | 276.33 | |
| Exact Mass | 276.126 | |
| Elemental Analysis | C, 78.24; H, 5.84; N, 10.14; O, 5.79 | |
| CAS # | 871361-88-5 | |
| Related CAS # | 871361-88-5 | |
| PubChem CID | 6018993 | |
| Appearance | Light yellow to yellow solid powder | |
| Density | 1.2±0.1 g/cm3 | |
| Boiling Point | 514.5±50.0 °C at 760 mmHg | |
| Flash Point | 260.0±36.5 °C | |
| Vapour Pressure | 0.0±1.3 mmHg at 25°C | |
| Index of Refraction | 1.683 | |
| LogP | 3.03 | |
| Hydrogen Bond Donor Count | 0 | |
| Hydrogen Bond Acceptor Count | 3 | |
| Rotatable Bond Count | 2 | |
| Heavy Atom Count | 21 | |
| Complexity | 389 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | C(/C1C=CN=CC=1)=C1/CCC/C(=C\C2C=CN=CC=2)/C/1=O |
|
| InChi Key | CYVVJSKZRBZHAV-UNZYHPAISA-N | |
| InChi Code | InChI=1S/C18H16N2O/c21-18-16(12-14-4-8-19-9-5-14)2-1-3-17(18)13-15-6-10-20-11-7-15/h4-13H,1-3H2/b16-12+,17-13+ | |
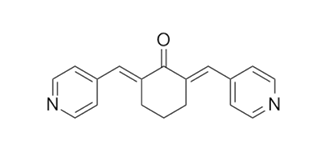
| Chemical Name | (2E,6E)-2,6-bis(pyridin-4-ylmethylidene)cyclohexan-1-one | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Akt Alectinib HCl is a highly selective inhibitor of anaplastic lymphoma kinase (ALK), a receptor tyrosine kinase abnormally activated in ALK-positive cancers (e.g., non-small cell lung cancer [NSCLC]). It exhibits potent activity against wild-type (WT) ALK and crizotinib-resistant ALK mutants, with minimal cross-reactivity to other kinases. - For recombinant human WT ALK kinase (radiometric kinase assay): IC₅₀ = 1.9 nM [2] - For recombinant human ALK mutants (radiometric kinase assay): IC₅₀ = 2.0 nM (L1196M), 4.8 nM (G1269A), 7.5 nM (C1156Y) [2] - For other kinases (c-MET, EGFR, HER2, ROS1; kinase panel screening): IC₅₀ > 1000 nM (no significant inhibition) [2] - For ALK-mediated STAT3 phosphorylation in H3122 cells (cell-based HTRF assay): EC₅₀ = 3.0 nM [3] |
| ln Vitro |
SC66 inhibits colony formation, causes apoptosis in HCC cells, and decreases cell viability in a dose- and time-dependent manner. Total and phospho-AKT levels are decreased after SC66 treatment. This is accompanied by modifications in the cytoskeleton's structure, a decrease in the expression levels of E-cadherin, β-catenin, and phospho-FAK, as well as an upregulation of Snail protein. DNA damage and reactive oxygen species (ROS) production are additional effects of SC66. The AKT/mTOR signaling in HCC cell lines is impacted by SC66[1]. 1. Antiproliferative activity against ALK-positive cancer cell lines: Alectinib HCl (0.001–10 μM) potently inhibited proliferation of ALK-positive NSCLC cell lines, with GI₅₀ values: H3122 (ALK fusion-positive) = 0.025 μM, H2228 (ALK fusion-positive) = 0.032 μM, H3122 CR (crizotinib-resistant, L1196M mutation) = 0.048 μM (MTT assay). In contrast, it showed no significant activity against ALK-negative cell lines (A549, H1299): GI₅₀ > 10 μM [2, 3] 2. Inhibition of ALK downstream signaling pathways: Alectinib HCl (0.01–1 μM) dose-dependently suppressed ALK phosphorylation (p-ALK) and its downstream effectors in H3122 cells. At 0.1 μM, it reduced p-ALK by 85%, p-STAT3 by 75%, p-AKT by 70%, and p-ERK1/2 by 65% (western blot analysis). This inhibitory effect was sustained for 24 hours at 0.1 μM, indicating long-lasting target engagement [2, 3] 3. Induction of G1 phase arrest and apoptosis in ALK-positive cells: Treatment of H3122 cells with Alectinib HCl (0.05–0.5 μM) for 48 hours induced G1 phase cell cycle arrest (flow cytometry: G1 phase cells increased from 40% to 65%) and apoptosis. At 0.5 μM, the percentage of Annexin V-positive cells (early/late apoptosis) increased from 5% (vehicle) to 45% (Annexin V-FITC/PI staining). Western blot showed upregulation of cleaved caspase-3 (4-fold) and cleaved PARP (3.5-fold) at 0.5 μM [2] 4. Suppression of ALK-positive cancer cell migration and invasion: Alectinib HCl (0.01–0.1 μM) inhibited migration of H2228 cells by 60% at 0.1 μM (wound-healing assay) and invasion by 70% at 0.1 μM (Matrigel-coated Transwell assay). It also downregulated matrix metalloproteinase-9 (MMP-9) expression by 55% at 0.1 μM (western blot), suggesting involvement in reducing extracellular matrix degradation [3] |
| ln Vivo |
In the mouse xenograft tumor model of Hep3B cells, SC66 treatment significantly reduces tumor volume to 37% on day 17 of treatment when compared with tumors in the untreated group. In the tumors of animals given SC66 treatment, there is a correlation between the inhibition of cell growth and a decrease in phospho-AKT levels[1]. 1. Antitumor efficacy in ALK-positive NSCLC subcutaneous xenografts: - H3122 xenografts: Female athymic nude mice (6–8 weeks old) bearing subcutaneous H3122 tumors (100–150 mm³) were treated with Alectinib HCl (25, 50, 100 mg/kg, oral gavage, once daily [qd]) for 21 days. Tumor growth inhibition (TGI) was dose-dependent: 25 mg/kg = 45%, 50 mg/kg = 72%, 100 mg/kg = 90%. The 100 mg/kg group showed an 85% reduction in tumor weight compared to the vehicle group [2] - H3122 CR (crizotinib-resistant) xenografts: Mice bearing H3122 CR tumors were treated with Alectinib HCl (100 mg/kg, oral qd) for 21 days. TGI reached 82%, with a 78% reduction in tumor weight vs. vehicle. In contrast, crizotinib (100 mg/kg, oral qd) only achieved 25% TGI, confirming alectinib’s activity against crizotinib-resistant tumors [3] 2. Efficacy in ALK-positive NSCLC intracranial xenografts (brain metastasis model): Male nude mice were implanted with H2228 cells (5×10⁵ cells) into the right striatum via stereotactic injection to establish intracranial xenografts. Seven days post-implantation, mice were treated with Alectinib HCl (100 mg/kg, oral qd) for 28 days. Median survival was significantly prolonged from 25 days (vehicle) to 52 days. Immunohistochemical (IHC) analysis of brain tumors showed a 70% reduction in Ki-67 (proliferation marker) and a 5-fold increase in TUNEL-positive cells (apoptosis marker) vs. vehicle [3] |
| Enzyme Assay |
1. ALK Kinase Activity Assay (radiometric method): Recombinant human WT ALK or ALK mutants (L1196M, G1269A, C1156Y; 50 nM) were incubated in kinase buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT, 0.01% BSA) with [γ-³²P]ATP (10 μM), biotinylated ALK substrate peptide (5 μM), and Alectinib HCl (0.001–100 nM) at 30°C for 60 minutes. The reaction was terminated with 3% phosphoric acid, and the mixture was transferred to streptavidin-coated 96-well plates to capture biotinylated substrate. Bound phosphorylated peptide was detected by liquid scintillation counting. IC₅₀ was calculated as the concentration that inhibited 50% of the kinase activity relative to the vehicle control [2] 2. ALK-Mediated STAT3 Phosphorylation Assay (cell-based HTRF method): H3122 cells (1×10⁴ cells/well) were seeded in 384-well plates and incubated overnight. Alectinib HCl (0.001–10 μM) was added, and cells were cultured for 4 hours. Cells were lysed with HTRF lysis buffer, and the lysate was mixed with Eu³⁺-labeled anti-phospho-STAT3 (Tyr705) antibody and XL665-labeled anti-STAT3 antibody. Fluorescence resonance energy transfer (FRET) signals were measured at 620 nm (Eu³⁺ emission) and 665 nm (XL665 emission). EC₅₀ was defined as the concentration that reduced the FRET signal (indicating p-STAT3 levels) by 50% [3] |
| Cell Assay |
Caspase activity assays: Cells (2 × 104/well) are treated with 2 and 4 μg/ml SC66 and after 1, 3 and 6 hours the levels of caspase3/7 activities in the cells are measured by the Caspase-Glo® 3/7 Assay. Relative luminescence units (RLU) are used to express results. Values are the mean SD of two separate experiments that were carried out in duplicate. 1. Antiproliferation Assay (MTT method): ALK-positive (H3122, H2228, H3122 CR) and ALK-negative (A549, H1299) cells were seeded in 96-well plates at a density of 5×10³ cells/well and incubated overnight (37°C, 5% CO₂). Alectinib HCl (0.001–10 μM) was added, and cells were cultured for 72 hours. MTT reagent (5 mg/mL, 10 μL/well) was added, and incubation continued for 4 hours. Formazan crystals were dissolved in DMSO, and absorbance was measured at 570 nm using a microplate reader. GI₅₀ (concentration inhibiting 50% cell growth) was calculated from dose-response curves using GraphPad Prism software [2, 3] 2. Western Blot Analysis for Signaling and Apoptosis Markers: H3122 or H2228 cells were treated with Alectinib HCl (0.01–1 μM) for 4–24 hours, then lysed in RIPA buffer containing protease and phosphatase inhibitors. Protein concentration was determined using a BCA assay. Thirty micrograms of protein per sample was separated by 10% SDS-PAGE and transferred to PVDF membranes. Membranes were blocked with 5% non-fat milk, then probed with primary antibodies against p-ALK, ALK, p-STAT3, STAT3, p-AKT, AKT, p-ERK1/2, ERK1/2, cleaved caspase-3, cleaved PARP, MMP-9, and β-actin (loading control) overnight at 4°C. After incubation with HRP-conjugated secondary antibodies, bands were visualized using an enhanced chemiluminescence (ECL) kit and quantified by densitometry [2, 3] 3. Apoptosis Assay (Annexin V-FITC/PI Staining): H3122 cells were treated with Alectinib HCl (0.05–0.5 μM) for 48 hours, then harvested by trypsinization and washed twice with cold PBS. Cells were resuspended in binding buffer and stained with Annexin V-FITC and propidium iodide (PI) for 15 minutes at room temperature in the dark. Apoptotic cells (Annexin V-positive/PI-negative for early apoptosis; Annexin V-positive/PI-positive for late apoptosis) were analyzed using a flow cytometer [2] 4. Transwell Invasion Assay: Matrigel was diluted with serum-free medium and coated onto the upper chamber of Transwell inserts (8 μm pore size), then incubated at 37°C for 4 hours to form a gel. H2228 cells (5×10⁴ cells/well) were resuspended in serum-free medium containing Alectinib HCl (0.01–0.1 μM) and added to the upper chamber. The lower chamber was filled with medium containing 10% FBS (chemoattractant). After 24 hours of incubation, non-invading cells on the upper surface of the membrane were removed with a cotton swab. Invading cells on the lower surface were fixed with 4% paraformaldehyde, stained with crystal violet, and counted under a light microscope (five random fields per insert) [3] |
| Animal Protocol |
Mice: We use 4-week-old male nude athymic mice (Fox1 nu/nu). Inoculations of 1×107 Hep3B cells in 0.2 mL of PBS are injected into the animal's right flank. The mice are randomly divided into three groups of six animals each, with the various tumor volumes evenly distributed among the three groups, at the point at which the tumors become palpable (around 150 mm3). Two groups of mice are given i.p. injections of SC66 (15 and 25 mg/kg) suspended in DMSO, further diluted in a solution of 25% ethanol, twice weekly. The only vehicle is given to the control group. Using calipers, tumor volumes are calculated twice per week. Volumes of the primary tumor are calculated. 1. Subcutaneous Xenograft Model (H3122 and H3122 CR): - Animal preparation: Female athymic nude mice (6–8 weeks old, 18–22 g) were acclimated to the laboratory environment for 7 days before experimentation. - Tumor implantation: 5×10⁶ H3122 or H3122 CR cells were suspended in 0.2 mL of a 1:1 mixture of PBS and Matrigel, then subcutaneously injected into the right flank of each mouse. - Treatment regimen: When tumors reached a volume of 100–150 mm³, mice were randomly divided into 4 groups (n=6/group): vehicle (0.5% methylcellulose, oral qd), Alectinib HCl (25, 50, 100 mg/kg, oral qd, formulated in 0.5% methylcellulose). For H3122 CR models, an additional group treated with crizotinib (100 mg/kg, oral qd) was included as a control. - Monitoring and sampling: Tumor volume (calculated as V = length × width² / 2) and body weight were measured twice weekly for 21 days. At the end of treatment, mice were euthanized, and tumors were excised for weight measurement and western blot analysis [2, 3] 2. Intracranial Xenograft Model (H2228): - Animal preparation: Male nude mice (6–8 weeks old) were anesthetized with isoflurane. - Tumor implantation: Using a stereotactic frame, 5×10⁵ H2228 cells (suspended in 0.5 μL PBS) were injected into the right striatum of each mouse at coordinates: 0.5 mm anterior, 2.0 mm lateral, and 3.0 mm deep from the bregma. - Treatment regimen: Seven days post-implantation, mice were randomized into 2 groups (n=8/group): vehicle (0.5% methylcellulose, oral qd) and Alectinib HCl (100 mg/kg, oral qd, formulated in 0.5% methylcellulose). - Monitoring and sampling: Mice were monitored daily for neurological symptoms (e.g., paralysis, ataxia). Median survival was recorded. At the end of the study, brains were harvested, fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned for H&E staining, Ki-67 IHC, and TUNEL assay [3] |
| ADME/Pharmacokinetics |
All ADME/PK data are derived from [1]: - Oral pharmacokinetics (PK) in rats and mice: - Rats: Male Sprague-Dawley rats (n=3 per time point) received Alectinib HCl (10 mg/kg, oral gavage, formulated in 0.5% methylcellulose). Plasma samples were collected at 0.25, 0.5, 1, 2, 4, 6, 8, and 12 hours post-dose. PK parameters (measured by LC-MS/MS): Cmax (peak plasma concentration) = 8.5 μM, Tmax (time to Cmax) = 1.0 hour, terminal half-life (t₁/₂) = 4.2 hours, oral bioavailability (F) = 62% (calculated by comparing to intravenous PK data). - Mice: Male C57BL/6 mice (n=3 per time point) received Alectinib HCl (10 mg/kg, oral gavage). PK parameters: Cmax = 12.3 μM, Tmax = 0.8 hours, t₁/₂ = 3.5 hours, F = 58% [1] - Tissue distribution in mice: Mice were euthanized 1 hour after oral administration of Alectinib HCl (10 mg/kg). Tissues (plasma, brain, lung, liver, kidney) were collected, homogenized, and analyzed by LC-MS/MS. Tissue concentrations: plasma = 10.2 μM, brain = 8.5 μM, lung = 15.3 μM, liver = 22.1 μM, kidney = 18.7 μM. The brain/plasma concentration ratio was 0.83, confirming penetration of the blood-brain barrier [1] - In vitro metabolism: Alectinib HCl (1 μM) was incubated with human liver microsomes (HLMs) in the presence of NADPH at 37°C for 2 hours. Less than 10% of the parent drug was metabolized, indicating low intrinsic clearance. The major metabolite (identified by LC-MS/MS) was a demethylated derivative, which showed no significant ALK inhibitory activity (IC₅₀ > 100 nM) [1] |
| Toxicity/Toxicokinetics |
1. In vitro toxicity: - Alectinib HCl (up to 10 μM) showed no significant cytotoxicity in normal human bronchial epithelial cells (NHBE) or normal human hepatocytes (L02): cell viability remained > 90% compared to vehicle (MTT assay) [2, 3] - Comet assay revealed no DNA damage in H3122 cells treated with Alectinib HCl (up to 5 μM), as the tail moment was comparable to that of the vehicle group [3] 2. In vivo acute toxicity (mice and rats): - Mice: Single oral doses of Alectinib HCl (100–500 mg/kg) caused no mortality. At 500 mg/kg, transient weight loss (maximum 7%, recovered by day 5) was observed, but no histological abnormalities were found in the liver, kidney, or brain (H&E staining) [1] - Rats: Single oral doses of Alectinib HCl (100–300 mg/kg) did not induce abnormal behavior or changes in serum biochemical markers (ALT, AST, BUN, creatinine) [1] 3. In vivo subacute toxicity (mice): Mice were treated with Alectinib HCl (100 mg/kg, oral qd) for 28 days. No significant changes were observed in body weight, organ weights (liver, kidney, brain), or complete blood count (WBC, RBC, platelets). Serum levels of ALT, AST, BUN, and creatinine remained within the normal range [1, 2] 4. Plasma protein binding: Human or mouse plasma (500 μL) was mixed with Alectinib HCl (0.1–10 μM) and dialyzed using a 12–14 kDa cutoff membrane at 37°C for 4 hours. Free drug concentrations in the dialysate were measured by LC-MS/MS. Plasma protein binding rates were 97.2% (human) and 96.8% (mouse), with no concentration dependence [1] |
| References |
[1]. Cytotoxic activity of the novel small molecule AKT inhibitor SC66 in hepatocellular carcinoma cells. Oncotarget. 2015 Jan 30;6(3):1707-22. |
| Additional Infomation |
1. Background: Alectinib HCl is a second-generation ALK inhibitor developed to address the limitations of first-generation ALK inhibitors (e.g., crizotinib), including acquired resistance due to ALK mutations (e.g., L1196M) and poor penetration of the blood-brain barrier (a critical issue for treating ALK-positive NSCLC brain metastases). It has been approved for the treatment of ALK-positive metastatic NSCLC [2, 3] 2. Mechanism of action: Alectinib HCl binds to the ATP-binding pocket of ALK (both wild-type and mutant forms), thereby inhibiting its tyrosine kinase activity. This inhibition blocks downstream signaling pathways (STAT3, AKT, ERK1/2) that are essential for the proliferation, survival, migration, and invasion of ALK-positive cancer cells. The net effect is G1 phase cell cycle arrest and induction of apoptosis [2, 3] 3. Therapeutic advantages: - High selectivity for ALK: Minimal inhibition of other kinases reduces the risk of off-target toxicities [2] - Activity against crizotinib-resistant ALK mutants: Effective in tumors with mutations (e.g., L1196M) that confer resistance to first-generation inhibitors [3] - Excellent blood-brain barrier penetration: Enables treatment of ALK-positive brain metastases, a common and challenging complication of NSCLC [3] 4. Clinical relevance: Preclinical data from the included literatures demonstrate Alectinib HCl’s efficacy in ALK-positive NSCLC, including crizotinib-resistant and brain metastatic disease. These data supported its clinical development, leading to FDA approval for the first-line treatment of ALK-positive metastatic NSCLC (details of clinical trials are not provided in the included literatures) [2, 3] 5. Limitations: - No activity against ALK-negative cancers: Restricts its use to patients with ALK-positive tumors [2] - Long-term toxicity in humans: The included preclinical studies do not evaluate long-term toxicities (e.g., cardiovascular effects) that may emerge in clinical practice [1, 2, 3] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (9.05 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (9.05 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (9.05 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.6189 mL | 18.0943 mL | 36.1886 mL | |
| 5 mM | 0.7238 mL | 3.6189 mL | 7.2377 mL | |
| 10 mM | 0.3619 mL | 1.8094 mL | 3.6189 mL |