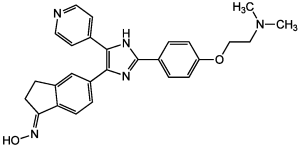
SB590885 (SB 590885; SB-590885) is a novel and potent B-Raf inhibitor with potential antineoplastic activity. With a Ki of 0.16 nM in a cell-free assay, it inhibits B-Raf and shows an 11-fold preference for B-Raf over c-Raf. It doesn't block kinases that are related to each other closely. In order to stabilize B-Raf'sactiveconformation, it is discovered that SB590885 binds to B-Raf inside the ATP-binding pocket. Only tumor cells expressing oncogenic B-Raf V600E are inhibited by SB590885's ability to phosphorylate ERK and show anti-proliferation. When exposed to SB-590885, cancer cell lines and normal cells exhibit varying sensitivity to or resistance to the treatment, whereas malignant cells expressing oncogenic B-Raf exhibit selective inhibition of mitogen-activated protein kinase activation, proliferation, transformation, and tumorigenicity.
Physicochemical Properties
| Molecular Formula | C27H27N5O2 | |
| Molecular Weight | 453.54 | |
| Exact Mass | 453.216 | |
| Elemental Analysis | C, 71.50; H, 6.00; N, 15.44; O, 7.06 | |
| CAS # | 405554-55-4 | |
| Related CAS # |
|
|
| PubChem CID | 135421339 | |
| Appearance | White to yellow solid | |
| Density | 1.3±0.1 g/cm3 | |
| Boiling Point | 691.1±55.0 °C at 760 mmHg | |
| Flash Point | 371.8±31.5 °C | |
| Vapour Pressure | 0.0±2.3 mmHg at 25°C | |
| Index of Refraction | 1.666 | |
| LogP | 5.01 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 6 | |
| Rotatable Bond Count | 7 | |
| Heavy Atom Count | 34 | |
| Complexity | 674 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | O([H])/N=C1/C2C([H])=C([H])C(C3=C(C4C([H])=C([H])N=C([H])C=4[H])N([H])C(C4C([H])=C([H])C(=C([H])C=4[H])OC([H])([H])C([H])([H])N(C([H])([H])[H])C([H])([H])[H])=N3)=C([H])C=2C([H])([H])C/1([H])[H] |
|
| InChi Key | MLSAQOINCGAULQ-QFMPWRQOSA-N | |
| InChi Code | InChI=1S/C27H27N5O2/c1-32(2)15-16-34-22-7-3-19(4-8-22)27-29-25(18-11-13-28-14-12-18)26(30-27)21-5-9-23-20(17-21)6-10-24(23)31-33/h3-5,7-9,11-14,17,33H,6,10,15-16H2,1-2H3,(H,29,30)/b31-24+ | |
| Chemical Name | (NE)-N-[5-[2-[4-[2-(dimethylamino)ethoxy]phenyl]-5-pyridin-4-yl-1H-imidazol-4-yl]-2,3-dihydroinden-1-ylidene]hydroxylamine | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
B-Raf (Ki = 0.16 nM); c-Raf (Ki = 1.72 nM) SB590885 is a selective inhibitor of oncogenic mutant B-Raf kinase (BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ). In recombinant human BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ kinase assays, it exhibits an IC₅₀ of 6 nM; it has low activity against wild-type BRAF (BRAFʷᵗ, IC₅₀ = 450 nM) and CRAF (IC₅₀ = 320 nM) [2] - In human BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ-positive A375 melanoma cells, SB590885 inhibits phosphorylated ERK1/2 (p-ERK, downstream of BRAF) with an EC₅₀ of 40 nM [1] - SB590885 shows no significant inhibition of non-RAF kinases, including MEK1 (IC₅₀ > 1 μM), EGFR (IC₅₀ > 5 μM), and PDGFRβ (IC₅₀ > 10 μM) [2] |
| ln Vitro |
SB590885 displays significant selectivity for B-Raf over c-Raf with Ki of 0.16 nM over 1.72 nM. The previously described Raf/VEGFR kinase inhibitor BAY 439006 (Ki = 38 nM for mutant B-Raf, 6 nM for c-Raf) is not as effective as SB-590885 as an inhibitor. Over 46 different kinases, SB590885 exhibits strong selectivity. The multi-kinase inhibitor BAY43-9006 does not stabilize the oncogenic B-Raf kinase domain in an active state, as does SB590885. SB590885 treatment potently inhibits ERK phosphorylation with EC50 values of 28 nM, 58 nM, 290 nM, 58 nM, and 190 nM, respectively, in Colo205, HT29, A375P, SKMEL28, and MALME-3M cells expressing oncogenic B-RafV600E. It also consistently inhibits proliferation with EC50 values of 0.1 μM, 0.87 μM, 0.37 μM, 0.12 μM, and 0.15 μM. SB590885 selectively inhibits the anchorage-independent growth of melanoma cell lines. [1] With a Kd of 0.3 nM, SB590885 exhibits high affinity for B-Raf. [2] The majority of melanoma cell lines (451Lu, WM35, and WM983) with the BRAF V600E mutation and no CDK4 mutations are very sensitive to SB590885, with an IC50 of <1 μM. In B-Raf V600E-mutated melanomas, elevated cyclin D1 levels brought on by genomic amplification are responsible for the drug's resistance. [3] BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ Melanoma Cell Proliferation Inhibition: In human BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ-positive melanoma cell lines (A375, SK-MEL-28), SB590885 (0.001–10 μM) concentration-dependently inhibits proliferation: A375 cells (IC₅₀ = 30 nM), SK-MEL-28 cells (IC₅₀ = 50 nM). At 0.1 μM, it reduces p-ERK levels by 90% (Western blot) and p-MEK levels by 85% in A375 cells, blocking the MAPK pathway [1] - BRAFʷᵗ Cell Insensitivity: In human BRAFʷᵗ melanoma cells (SK-MEL-5) and normal human foreskin fibroblasts (NHFF), SB590885 (up to 10 μM) shows minimal antiproliferative activity (viability reduced by <15%), confirming its mutant BRAF selectivity [1] - Cyclin D1-Mediated Resistance: In A375 cells overexpressing cyclin D1 (BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ-positive), SB590885’s antiproliferative activity is reduced: IC₅₀ increases from 30 nM (parental cells) to 180 nM (cyclin D1-overexpressing cells). At 0.1 μM, colony formation is inhibited by only 30% (vs. 80% in parental cells), with no significant change in p-ERK inhibition (still 90%) [3] - Apoptosis Induction in Sensitive Cells: In parental A375 cells, SB590885 (0.1–0.5 μM) induces apoptosis in a concentration-dependent manner: 0.5 μM increases Annexin V⁺ cells from 4% (vehicle) to 35%, accompanied by cleavage of caspase-3 and PARP (Western blot) [1] |
| ln Vivo |
In murine xenografts created from mutant B-Raf-expressing A375P melanoma cells, SB-590885 administration significantly and modestly inhibits tumor growth[1]. Prolonged exposure to SB-590885 inhibits cellular transformation and proliferation in three-dimensional culture. A, SB-590885 inhibits anchorage-independent tumor colony growth selectively in tumor cell lines expressing B-RafV600E (light microscopy, 4×). B, anchorage-independent growth was quantified by counting individual colonies on a grid in triplicate and normalizing to vehicle controls (±SE) for each cell line. C, spheroid cultures of HT-1080, WM-NCI, and WM-3434 were treated with a range of SB-590885 for 72 hours, and assessed for invasiveness by light microscopy (top) and metabolic activity by using the “Live/Dead cell assay” as indicated by the green and red fluorescent images, respectively. D, decreased tumorigenicity of A375P cells following treatment with SB-590885. Points, tumor growth for cohorts of animals treated with vehicle alone (▪) or with SB-590885 (X); bars, ±SE. Mice were treated for 21 days, and following this, the mice treated with SB-590885 were observed for an additional 14 days.[1] A375 Melanoma Xenograft Model: In female nude mice bearing A375 BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ xenografts, oral SB590885 (50, 100 mg/kg/day, once daily) dose-dependently inhibits tumor growth: 100 mg/kg reduces tumor volume by 70% at day 21 vs. vehicle, with no significant weight loss (<5%). Tumor p-ERK levels are reduced by 80% (immunohistochemistry), and tumor cell apoptosis is increased by 3-fold (TUNEL staining) [1] - BRAFʷᵗ Tumor Insensitivity: In nude mice bearing BRAFʷᵗ SK-MEL-5 xenografts, oral SB590885 (100 mg/kg/day) shows no significant tumor inhibition (volume reduced by <10%), consistent with its in vitro selectivity [1] |
| Enzyme Assay |
SB-590885 is a potent B-Raf inhibitor with Ki of 0.16 nM, and has 11-fold greater selectivity for B-Raf over c-Raf, without inhibition to other human kinases. In summary, SB-590885 (33), a potent and extremely selective inhibitor of B-Raf kinase, was identified following an evaluation of the SAR of a series of imidazole based leads. SB-590885 represents an excellent molecule with which to investigate the role of B-Raf in neurodegenerative and other disease states.[2] Compound activity is determined as either the ability of test compounds to inhibit B-Raf mediated phosphorylation of kinase dead MEK (data expressed as IC50) or as a B-Raf-binding affinity derived from the fluorescent ligand displacement assay (FP, data expressed as Kd). For assay details, see Dean, D. K.; Takle, A. K.; Wilson, D. M. PCT Int. Appl. WO 02/24680. Oncogenic BRAF alleles are both necessary and sufficient for cellular transformation, suggesting that chemical inhibition of the activated mutant protein kinase may reverse the tumor phenotype. Here, we report the characterization of SB-590885, a novel triarylimidazole that selectively inhibits Raf kinases with more potency towards B-Raf than c-Raf. Crystallographic analysis revealed that SB-590885 stabilizes the oncogenic B-Raf kinase domain in an active configuration, which is distinct from the previously reported mechanism of action of the multi-kinase inhibitor, BAY43-9006. Malignant cells expressing oncogenic B-Raf show selective inhibition of mitogen-activated protein kinase activation, proliferation, transformation, and tumorigenicity when exposed to SB-590885, whereas other cancer cell lines and normal cells display variable sensitivities or resistance to similar treatment. These studies support the validation of oncogenic B-Raf as a target for cancer therapy and provide the first evidence of a correlation between the expression of oncogenic BRAF alleles and a positive response to a selective B-Raf inhibitor.[1] Recombinant BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ Kinase Assay: Recombinant human BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ, BRAFʷᵗ, or CRAF protein (50 ng/well) was incubated in kinase buffer (25 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT, 20 μM ATP) with a biotinylated MEK1-derived peptide (substrate, 2 μM) and various concentrations of SB590885 (0.001–100 μM) at 30°C for 60 min. Phosphorylated substrate was detected using a homogeneous time-resolved fluorescence (HTRF) assay (Eu-labeled anti-phospho-MEK antibody + streptavidin-allophycocyanin). Kinase activity was normalized to vehicle control, and IC₅₀ values were calculated via nonlinear regression [2] |
| Cell Assay |
Adherent Cell Proliferation Analysis[3] Cells were plated into a 96-well plate at a density of 2.5 × 104 cells per milliliter and left to grow overnight. Cells were treated with increasing concentrations of SB-590885 in triplicate, after 72 h, the levels of growth inhibition were examined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Data show the mean of at least three independent experiments ± SE. To determine the role of cyclin D1 and CDK4 overexpression in resistance to SB-590885, 451Lu cells were infected with virus for either cyclin D1 or CDK4. CDK4-overexpressing cells were selected for following puromycin treatment for 48 h. After infection was confirmed by Western blotting, cells were plated out for the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay as described above.[3] Cell Cycle Analysis[3] Cells were plated into 10 cm dishes at 60% confluency and left to grow overnight before being treated with SB-590885 (1 and 3 μmol/L) for 24 h. Cells were ethanol-fixed, stained with propidium iodide, and analyzed as previously described. In some experiments, cells were infected with virus for either CDK4, cyclin D1, or CDK4 + cyclin D1 prior to treatment with SB-590885 and cell cycle analysis. [3] For the proliferation tests, cells are treated with substances in 0.1% DMSO and incubated for 72 hours at 37°C with 5% CO2. A Victor 2V plate reader and the CellTiter-Glo reagent are used to measure the number of viable cells. According to the directions provided by the manufacturer, cells are prepared for cell cycle analysis on a Becton Dickinson FACScan. Using the software CellQuest v3.3, data is gathered and examined. Using inhibitors or DMSO vehicle in the agar layer, anchorage-independent growth assays are carried out as previously described. For a total of 28 days, cultures are fed again with media, an inhibitor, or DMSO every 5 to 7 days. Utilizing standard light microscopy, colonies are viewed, captured on camera, and quantified by triplicate counting on a grid. Melanoma Cell Proliferation Assay: A375/SK-MEL-28/SK-MEL-5/NHFF cells were seeded in 96-well plates (5×10³ cells/well) in DMEM + 10% FBS. After 24 h adhesion, SB590885 (0.001–10 μM) was added, and cells were incubated for 72 h. Cell viability was measured via MTT assay (absorbance at 570 nm), and IC₅₀ values were determined using GraphPad Prism [1] - p-ERK/p-MEK Western Blot Assay: A375 cells (parental or cyclin D1-overexpressing) were seeded in 6-well plates (2×10⁵ cells/well) and treated with SB590885 (0.01–0.5 μM) for 24 h. Cells were lysed in RIPA buffer with protease/phosphatase inhibitors, and lysates (20 μg protein/lane) were separated by SDS-PAGE. Membranes were probed with anti-p-ERK, anti-total ERK, anti-p-MEK, anti-total MEK, anti-cleaved caspase-3, or anti-PARP antibodies, followed by HRP-conjugated secondary antibodies. Bands were visualized via chemiluminescence [1,3] - Colony Formation Assay: Cyclin D1-overexpressing A375 cells and parental cells were seeded in 6-well plates (1×10³ cells/well) and treated with SB590885 (0.01–0.5 μM) for 14 days. Colonies were fixed with methanol, stained with crystal violet, and counted. Colony formation inhibition was calculated relative to vehicle control [3] - Apoptosis Assay: Parental A375 cells were treated with SB590885 (0.1–0.5 μM) for 48 h, harvested, and stained with Annexin V-FITC and propidium iodide (PI). Apoptotic cells (Annexin V⁺/PI⁻ and Annexin V⁺/PI⁺) were quantified via flow cytometry [1] |
| Animal Protocol |
Following i.p. injection, the pharmacokinetic characteristics and safety of SB-590885 were assessed. It was discovered that daily injections of 50 mg/kg resulted in therapeutic levels with only minor changes in body weight. 8 to 12-week-old female nude mice are used to create the tumors. Tumors are induced by injecting 5×106 A375P cells in Matrigel suspension intravenously. Three weeks later, when the tumors have reached a volume of 150 to 250 mm3, the mice are randomized into groups of eight and then given the appropriate treatment. Animals are administered either the vehicle (2% N,N-dimethylacetamide, 2% Cremophor EL, and 96% acidified water (pH 4-5)) or the vehicle (daily for 21 days) containing 50 mg/kg of SB-590885. Then, 14 days after the end of the treatment, a cohort of mice that had been given SB-590885 are observed once more. Two times per week, calipers are used to measure the tumor volume for 55 days. A375/SK-MEL-5 Xenograft Protocol: Female nude mice (6–7 weeks old, 18–22 g) were subcutaneously injected with A375 (5×10⁶ cells/mouse) or SK-MEL-5 (1×10⁷ cells/mouse) in Matrigel (1:1 v/v) into the right flank. When tumors reached 100–120 mm³, mice were randomized into 3 groups (n=7/group): Vehicle (0.5% methylcellulose + 0.1% Tween 80, p.o.), SB590885 50 mg/kg (p.o., q.d.), SB590885 100 mg/kg (p.o., q.d.). Drugs were administered daily for 21 days. Tumor volume (V = π×L×W²/6) and body weight were measured every 3 days. At study end, tumors were excised: half were fixed in formalin for p-ERK immunohistochemistry and TUNEL staining; half were frozen for protein extraction [1] |
| Toxicity/Toxicokinetics |
Plasma Protein Binding: In mouse plasma (measured via ultrafiltration), SB590885 has a protein binding rate of ~90% at concentrations of 0.01–1 μM, with no concentration dependence [1] - Acute Toxicity: In nude mice treated with SB590885 (up to 100 mg/kg/day for 21 days), no mortality or severe toxicity is observed. Body weight remains stable, and serum ALT/AST (liver markers) and creatinine (renal marker) are within normal ranges [1] - Normal Tissue Safety: In NHFF cells (normal fibroblasts), SB590885 (up to 10 μM) shows no significant cytotoxicity (viability reduced by <10%), indicating a favorable therapeutic index [1] |
| References |
[1]. Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885. Cancer Res, 2006, 66(23), 11100-11105. [2]. The identification of potent and selective imidazole-based inhibitors of B-Raf kinase. Bioorg Med Chem Lett, 2006, 16(2), 378-381. [3]. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther, 2008, 7(9), 2876-2883. |
| Additional Infomation |
(E)-SB-590885 is an N-{5-[2-{4-[2-(dimethylamino)ethoxy]phenyl}-4-(pyridin-4-yl)-1H-imidazol-5-yl]-2,3-dihydro-1H-inden-1-ylidene}hydroxylamine in which the oxime group has E configuration. Recent studies have shown that there is a considerable heterogeneity in the response of melanoma cell lines to MEK and BRAF inhibitors. In the current study, we address whether dysregulation of cyclin-dependent kinase 4 (CDK4) and/or cyclin D1 contribute to the BRAF inhibitor resistance of melanoma cells. Mutational screening identified a panel of melanoma cell lines that harbored both a BRAF V600E mutation and a CDK4 mutation: K22Q (1205Lu), R24C (WM39, WM46, and SK-Mel-28), and R24L (WM902B). Pharmacologic studies showed that the presence of a CDK4 mutation did not alter the sensitivity of these cell lines to the BRAF inhibitor. The only cell line with significant BRAF inhibitor resistance was found to harbor both a CDK4 mutation and a CCND1 amplification. Array comparative genomic hybridization analysis showed that CCND1 was amplified in 17% of BRAF V600E-mutated human metastatic melanoma samples, indicating the clinical relevance of this finding. As the levels of CCND1 amplification in cell lines are lower than those seen in clinical specimens, we overexpressed cyclin D1 alone and in the presence of CDK4 in a drug-sensitive melanoma line. Cyclin D1 overexpression alone increased resistance and this was enhanced when cyclin D1 and CDK4 were concurrently overexpressed. In conclusion, increased levels of cyclin D1, resulting from genomic amplification, may contribute to the BRAF inhibitor resistance of BRAF V600E-mutated melanomas, particularly when found in the context of a CDK4 mutation/overexpression.[3] SB590885 is a research-grade selective BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ inhibitor, developed to validate the therapeutic potential of targeting mutant BRAF in cancers with BRAF V600E mutations. It is not approved for clinical use [1,2] - Mechanism of Action: Its antitumor effect is mediated by specific inhibition of BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ kinase activity, blocking the downstream MAPK (RAF-MEK-ERK) signaling pathway—this pathway is constitutively activated in BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ-positive cancers, driving cell proliferation and survival [1,2] - Resistance Mechanism: Preclinical studies show SB590885 resistance in BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ melanoma cells can be mediated by cyclin D1 overexpression, which bypasses MAPK pathway inhibition to maintain cell cycle progression. This highlights the need for combination strategies (e.g., BRAF + cyclin D1 inhibitors) [3] - Research Significance: SB590885 was one of the early selective BRAFⁿᵉᵗ/ᵛ⁶⁰⁰ᴱ inhibitors, providing foundational evidence for the clinical development of later BRAF inhibitors (e.g., vemurafenib) [1,2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.51 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.51 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.51 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 2% Cremophor EL, 2% N,N-dimethylacetamide, pH 5.0: 30mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2049 mL | 11.0244 mL | 22.0488 mL | |
| 5 mM | 0.4410 mL | 2.2049 mL | 4.4098 mL | |
| 10 mM | 0.2205 mL | 1.1024 mL | 2.2049 mL |