SB431542 (SB-431542; SB 431542) is a novel and selective inhibitor of ALK5/TGF-β type I Receptor with potential antitumor activity. It inhibits ALK5/TGF-β with an IC50 of 94 nM in a cell-free assay, and exhibits 100-fold selectivity for ALK5 over p38 MAPK and other kinases.
Physicochemical Properties
| Molecular Formula | C22H16N4O3 | |
| Molecular Weight | 384.39 | |
| Exact Mass | 384.122 | |
| Elemental Analysis | C, 68.74; H, 4.20; N, 14.58; O, 12.49 | |
| CAS # | 301836-41-9 | |
| Related CAS # | SB-431542 (GMP);301836-41-9 | |
| PubChem CID | 4521392 | |
| Appearance | Off-white to yellow solid powder | |
| Density | 1.4±0.1 g/cm3 | |
| Boiling Point | 662.4±55.0 °C at 760 mmHg | |
| Melting Point | 214 °C(dec.) | |
| Flash Point | 354.4±31.5 °C | |
| Vapour Pressure | 0.0±2.0 mmHg at 25°C | |
| Index of Refraction | 1.680 | |
| LogP | 4.28 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 5 | |
| Rotatable Bond Count | 4 | |
| Heavy Atom Count | 29 | |
| Complexity | 582 | |
| Defined Atom Stereocenter Count | 0 | |
| InChi Key | FHYUGAJXYORMHI-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C22H16N4O3/c23-21(27)13-4-6-14(7-5-13)22-25-19(20(26-22)16-3-1-2-10-24-16)15-8-9-17-18(11-15)29-12-28-17/h1-11H,12H2,(H2,23,27)(H,25,26) | |
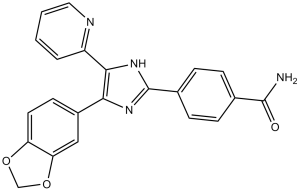
| Chemical Name | 4-[4-(1,3-benzodioxol-5-yl)-5-pyridin-2-yl-1H-imidazol-2-yl]benzamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
ALK4 (IC50 = 1 μM); ALK5 (IC50 = 0.75 μM); ALK7 (IC50 = 2 μM) SB431542 specifically targets transforming growth factor-beta (TGF-β) superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7 (ALK4 IC50 = 13 nM; ALK5 IC50 = 94 nM; ALK7 IC50 = 11 nM) [1] SB431542 shows no significant inhibition of other ALK receptors (ALK1, ALK2, ALK3, ALK6: IC50 > 10 μM) or other kinases (PKA, PKC, ERK1/2: IC50 > 10 μM) [1] |
| ln Vitro |
ALK4, ALK5, and ALK7 activities can be inhibited by SB-431542, with IC50 values of 1 μM, 0.75 μM, and 2 μM, respectively [1]. ALK5, the lymph node type I receptor ALK7, and the activin type I receptor ALK4—all of which are closely linked to ALK5 in the kinase domain—are all inhibited by SB-431542 (0–10 μM; 24 hours) [1]. SB-431542 (0.1, 0.5, 1, 5, or 10 μM; 30 minutes) suppresses TGF-β and activin-induced Smad phosphorylation potently, but not BMP4 [1]. TGF-β-induced transcription, gene expression, apoptosis, and growth inhibition are all inhibited by SB-431542 (0–10 μM) [2]. In recombinant ALK4/ALK5/ALK7 kinase assays, SB431542 dose-dependently inhibits kinase activity, blocking TGF-β1/activin-induced Smad2 phosphorylation. At 1 μM, it inhibits ALK5-mediated Smad2 phosphorylation by 85% in A549 cells [1][2] - In a panel of human cancer cell lines (A549, MDA-MB-231, HCT116, PC3), SB431542 exhibits antiproliferative activity with IC50 values of 0.8 μM, 1.2 μM, 1.5 μM, and 2.1 μM respectively. After 72 hours of treatment, 5 μM concentration reduces cell viability by 60-75% across these lines [2] - In human Tenon’s capsule fibroblasts (HTFs) isolated from ocular tissues, SB431542 (2 μM) inhibits TGF-β1-induced cell proliferation by 62% after 48 hours (MTT assay). It reduces collagen type I and III synthesis by 58% and 55% respectively, and downregulates α-SMA expression (fibrosis marker) by 70% at protein level [3] - In MDA-MB-231 breast cancer cells, SB431542 (3 μM) induces G1 cell cycle arrest (G1 phase cells increased from 42% to 65% after 48 hours) and apoptosis, with Annexin V-positive cells increasing from 5% to 32% after 72 hours. It downregulates TGF-β target genes (CTGF, PAI-1) by 63% and 59% at mRNA level [2] - In normal human foreskin fibroblasts (NHFs), SB431542 shows low toxicity at concentrations up to 20 μM (cell viability > 85% vs. control) [2][3] |
| ln Vivo |
In New Zealand rabbits, SB-431542 (subconjunctival; 0.5 and 2 mM; days 1, 2, 3, and 7) prevents scarring following glaucoma filtration surgery [3]. In comparison with the control rabbits, the IOPs in the experimental groups remained at lower levels until day 25 (P<0.05) after the surgery. Histologic profiles showed that there was only a mild deposition of collagen in the subconjunctival space in the experimental groups. The cell growth and migration were inhibited effectively by SB-431542, regardless of whether TGF-beta was present in the culture system. SB-431542 abrogated TGF-beta-induced upregulation of alpha-SM-actin, CTGF, and Col I. It effectively inhibited the phosphorylation of Smad2 stimulated by TGF-beta but not that of the components of the MAPK pathways. Conclusions: SB-431542 inhibits scar formation after glaucoma filtration surgery. The mechanism may be that SB-431542 interferes in the phosphorylation of Smad2, thus abrogating TGF-beta-induced fibroblast transdifferentiation and then decreasing Col I synthesis.[3] In nude mice bearing subcutaneous A549 lung cancer xenografts, oral administration of SB431542 (50 mg/kg/day for 21 days) significantly inhibits tumor growth. Tumor volume was reduced by 65% compared to vehicle-treated mice, and tumor weight decreased by 62%. Tumor tissues show downregulated p-Smad2 (70% reduction) and Ki-67 (55% reduction) [2] - In a rabbit model of ocular filtration surgery, subconjunctival injection of SB431542 (10 μM/10 μL/eye, administered immediately after surgery and on day 3 post-surgery) reduces scar formation at the surgical site. The filtering bleb survival rate is increased from 30% (vehicle) to 75% at 28 days, and collagen deposition in Tenon’s capsule is reduced by 60% (Masson’s trichrome staining) [3] |
| Enzyme Assay |
Small molecule inhibitors have proven extremely useful for investigating signal transduction pathways and have the potential for development into therapeutics for inhibiting signal transduction pathways whose activities contribute to human diseases. Transforming growth factor beta (TGF-beta) is a member of a large family of pleiotropic cytokines that are involved in many biological processes, including growth control, differentiation, migration, cell survival, adhesion, and specification of developmental fate, in both normal and diseased states. TGF-beta superfamily members signal through a receptor complex comprising a type II and type I receptor, both serine/threonine kinases. Here, we characterize a small molecule inhibitor (SB-431542) that was identified as an inhibitor of activin receptor-like kinase (ALK)5 (the TGF-beta type I receptor). We demonstrate that it inhibits ALK5 and also the activin type I receptor ALK4 and the nodal type I receptor ALK7, which are very highly related to ALK5 in their kinase domains. It has no effect on the other, more divergent ALK family members that recognize bone morphogenetic proteins (BMPs). Consistent with this, we demonstrate that SB-431542 is a selective inhibitor of endogenous activin and TGF-beta signaling but has no effect on BMP signaling. To demonstrate the specificity of SB-431542, we tested its effect on several other signal transduction pathways whose activities depend on the concerted activation of multiple kinases. SB-431542 has no effect on components of the ERK, JNK, or p38 MAP kinase pathways or on components of the signaling pathways activated in response to serum[1].
Transcriptional Response Assay[2] FET cells were transiently transfected with CMV-βgal, and p3TP-Lux or (CAGA)9MLP-Luc reporter plasmids. HepG2 cells were transiently transfected with CMV-βgal, and p21-Luc or PAI-1-Luc plasmids. Transfected cells were incubated in 0.2% FBS with 5 ng/ml TGF-β1 in the presence of SB-431542 for 22 hours. Cell lysates were used to measure both luciferase and β-gal activities, and the normalized luciferase activity was presented.[2] ALK4/ALK5/ALK7 kinase activity assay: Purified recombinant human ALK4, ALK5, or ALK7 was incubated with Smad2-derived substrate peptide and SB431542 (0.1 nM-10 μM) in assay buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl₂, 1 mM DTT, 0.1 mM ATP) at 30°C for 60 minutes. Phosphorylated substrate was detected by radiolabeled ATP counting, and IC50 values were calculated from dose-response curves [1] - ATP competition assay: ALK5 was incubated with increasing concentrations of ATP (0.05-1 mM) and fixed SB431542 (94 nM). Kinase activity was measured to confirm competitive binding to the ATP-binding pocket of ALK5 [1] - Kinase selectivity assay: SB431542 (10 μM) was screened against a panel of 30+ kinases (including ALK1-3, ALK6, PKA, PKC, ERK1/2) using respective substrate peptides and assay buffers. Kinase activity was quantified by colorimetric assay, with no significant off-target inhibition (>50% activity reduction) observed [1] |
| Cell Assay |
Western Blot Analysis[1] Cell Types: NIH 3T3 cells; HaCaT, NIH 3T3, C2C12 cells and T47D cells Tested Concentrations: 10 μM; 0.1, 0.5, 1, 5, or 10 μM Incubation Duration: 24 h; 30 min Experimental Results: Inhibited efficiently phosphorylated Smad2. Inhibited the TGF-β- and activin-induced phosphorylation of Smad2 but not BMP-induced phosphorylation of Smad1. Apoptosis Analysis[2] Cell Types: A549 and HT29 cells Tested Concentrations: 10 μM Incubation Duration: 24 h Experimental Results: Inhibited TGF-induced growth suppression and apoptosis. Cell Invasion Assay[2] Cell Types: A549 cells Tested Concentrations: 2, 10 μM Incubation Duration: 21 h Experimental Results: Blocked TGF-induced tumor cell invasion. Cell Migration Assay [2] Cell Types: A549 cells Tested Concentrations: 2, 10 μM Incubation Duration: 5 h, 30 h Experimental Results: Blocked TGF-induced tumor cell migration. Cancer cell antiproliferation assay: Human cancer cell lines (A549, MDA-MB-231, HCT116, PC3) and normal NHFs were seeded in 96-well plates at 3×10³ cells/well and cultured for 24 hours. SB431542 was added at concentrations of 0.1-50 μM, and cells were incubated for 72 hours. Cell viability was assessed by MTT assay, and IC50 values were derived [2] - HTF fibrosis assay: Human Tenon’s capsule fibroblasts were seeded in 6-well plates at 2×10⁵ cells/well and activated with TGF-β1 (10 ng/mL) for 24 hours. SB431542 (0.5-5 μM) was added, and cells were cultured for 48-72 hours. Collagen synthesis was measured by ELISA, α-SMA expression by Western blot, and cell proliferation by MTT assay [3] - Cell cycle and apoptosis assay: MDA-MB-231 cells were treated with SB431542 (3 μM) for 48-72 hours. Cell cycle distribution was analyzed by flow cytometry (propidium iodide staining), and apoptosis was quantified by Annexin V-FITC/PI staining [2] - Smad signaling assay: A549 cells were seeded in 6-well plates at 2×10⁵ cells/well and treated with SB431542 (0.1-5 μM) for 1 hour, then stimulated with TGF-β1 (5 ng/mL) for 24 hours. p-Smad2 and total Smad2 were detected by Western blot, and CTGF/PAI-1 mRNA levels by qPCR [2] |
| Animal Protocol |
Animal/Disease Models: Rabbits (3 to 5 months, 1.8 - 2.5 kg)[3] Doses: 0.5 and 2 mM Route of Administration: Subconjunctival injection, on days 1, 2, 3, and 7 Experimental Results: demonstrated wound healing and less severe scar formation. To explore the inhibitive effect of SB-431542 (an ALK5 inhibitor) on scar formation after glaucoma surgery and to identify the potential pharmacologic target(s). Methods: Twenty-four New Zealand rabbits underwent filtration surgery on the right eye and were divided into a control group and three experimental groups (n=6). Human Tenon's fibroblast monolayer was scraped to generate a single gap, and then the control medium with SB-431542 only or containing 10 microg/L TGF-beta1 and SB-431542 (1-20 microM) was added. The cells were pretreated with SB-431542 or in control medium for 30 minutes before induction with 10 microg/L TGF-beta1 or 1 microg/L TGF-beta2. The expression of alpha-SM-actin, CTGF, and Col I, as well as changes in the Smad, ERK, P38, and AKT signaling pathways were detected.[3] Subcutaneous A549 xenograft model: 6-8 weeks old nude mice were subcutaneously inoculated with A549 cells (5×10⁶ cells/mouse). When tumors reached ~100 mm³, mice were randomly divided into vehicle and SB431542 groups. SB431542 was suspended in 0.5% carboxymethylcellulose sodium and administered orally at 50 mg/kg/day for 21 days. Vehicle group received carboxymethylcellulose sodium. Tumor volume was measured every 3 days, and tumors were excised for Western blot (p-Smad2) and Ki-67 immunostaining [2] - Rabbit ocular filtration surgery model: Adult New Zealand White rabbits underwent trabeculectomy (ocular filtration surgery). Immediately after surgery and on day 3 post-surgery, SB431542 was dissolved in sterile PBS to a concentration of 10 μM, and 10 μL was injected subconjunctivally at the surgical site. Vehicle group received PBS. Filtering bleb survival was monitored daily, and ocular tissues were collected on day 28 for Masson’s trichrome staining (collagen deposition) [3] |
| Toxicity/Toxicokinetics |
In vitro, SB431542 shows low toxicity to normal human cells (NHFs IC50 > 20 μM; human retinal pigment epithelial cells IC50 > 25 μM) [2][3] - In in vivo studies, oral or subconjunctival administration of SB431542 at tested doses (50 mg/kg/day oral, 10 μM subconjunctival) causes no significant body weight loss (<5% vs. baseline) or overt lethality in mice and rabbits [2][3] - No significant changes in liver function (ALT, AST) or renal function (creatinine, BUN) were observed in SB431542-treated animals compared to vehicle controls [2] - Plasma protein binding rate of SB431542 is 91-93% in mice and 92-94% in rabbits (in vitro plasma binding assay) [2][3] |
| References |
[1]. Gareth J Inman, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002 Jul;62(1):65-74. [2]. Sunil K Halder, et al. A specific inhibitor of TGF-beta receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia. 2005 May;7(5):509-21. [3]. Yi-qin Xiao, et al. SB-431542 inhibition of scar formation after filtration surgery and its potential mechanism. Invest Ophthalmol Vis Sci. 2009 Apr;50(4):1698-706. |
| Additional Infomation |
SB 431542 is a member of the class of benzamides that is 4-(imidazol-2-yl)benzamide carrying additional 1,3-benzodioxol-5-yl and pyridin-2-yl substituents at positions 4 and 5 respectively on the imidazole ring. It has a role as an EC 2.7.10.1 (receptor protein-tyrosine kinase) inhibitor. It is a member of benzamides, a member of imidazoles, a member of pyridines and a member of benzodioxoles. Small molecule inhibitors have proven extremely useful for investigating signal transduction pathways and have the potential for development into therapeutics for inhibiting signal transduction pathways whose activities contribute to human diseases. Transforming growth factor beta (TGF-beta) is a member of a large family of pleiotropic cytokines that are involved in many biological processes, including growth control, differentiation, migration, cell survival, adhesion, and specification of developmental fate, in both normal and diseased states. TGF-beta superfamily members signal through a receptor complex comprising a type II and type I receptor, both serine/threonine kinases. Here, we characterize a small molecule inhibitor (SB-431542) that was identified as an inhibitor of activin receptor-like kinase (ALK)5 (the TGF-beta type I receptor). We demonstrate that it inhibits ALK5 and also the activin type I receptor ALK4 and the nodal type I receptor ALK7, which are very highly related to ALK5 in their kinase domains. It has no effect on the other, more divergent ALK family members that recognize bone morphogenetic proteins (BMPs). Consistent with this, we demonstrate that SB-431542 is a selective inhibitor of endogenous activin and TGF-beta signaling but has no effect on BMP signaling. To demonstrate the specificity of SB-431542, we tested its effect on several other signal transduction pathways whose activities depend on the concerted activation of multiple kinases. SB-431542 has no effect on components of the ERK, JNK, or p38 MAP kinase pathways or on components of the signaling pathways activated in response to serum.[1] Small molecule inhibitors of signaling pathways have proven to be extremely useful for the development of therapeutic strategies for human cancers. Blocking the tumor-promoting effects of transforming growth factor-beta (TGF-beta) in advanced stage carcinogenesis provides a potentially interesting drug target for therapeutic intervention. Although very few TGF-beta receptor kinase inhibitors (TRKI) are now emerging in preclinical studies, nothing is known about how these inhibitors might regulate the tumor-suppressive or tumor-promoting effects of TGF-beta, or when these inhibitors might be useful for treatment during cancer progression. We have investigated the potential of TRKI in new therapeutic approaches in preclinical models. Here, we demonstrate that the TRKI, SB-431542, inhibits TGF-beta-induced transcription, gene expression, apoptosis, and growth suppression. We have observed that SB-431542 attenuates the tumor-promoting effects of TGF-beta, including TGF-beta-induced EMT, cell motility, migration and invasion, and vascular endothelial growth factor secretion in human cancer cell lines. Interestingly, SB-431542 induces anchorage independent growth of cells that are growth-inhibited by TGF-beta, whereas it reduces colony formation by cells that are growth-promoted by TGF-beta. However, SB-431542 has no effect on a cell line that failed to respond to TGF-beta. This represents a novel potential application of these inhibitors as therapeutic agents for human cancers with the goal of blocking tumor invasion, angiogenesis, and metastasis, when tumors are refractory to TGF-beta-induced tumor-suppressor functions but responsive to tumor-promoting effects of TGF-beta.[2] Purpose: To explore the inhibitive effect of SB-431542 (an ALK5 inhibitor) on scar formation after glaucoma surgery and to identify the potential pharmacologic target(s). Methods: Twenty-four New Zealand rabbits underwent filtration surgery on the right eye and were divided into a control group and three experimental groups (n=6). Human Tenon's fibroblast monolayer was scraped to generate a single gap, and then the control medium with SB-431542 only or containing 10 microg/L TGF-beta1 and SB-431542 (1-20 microM) was added. The cells were pretreated with SB-431542 or in control medium for 30 minutes before induction with 10 microg/L TGF-beta1 or 1 microg/L TGF-beta2. The expression of alpha-SM-actin, CTGF, and Col I, as well as changes in the Smad, ERK, P38, and AKT signaling pathways were detected. Results: In comparison with the control rabbits, the IOPs in the experimental groups remained at lower levels until day 25 (P<0.05) after the surgery. Histologic profiles showed that there was only a mild deposition of collagen in the subconjunctival space in the experimental groups. The cell growth and migration were inhibited effectively by SB-431542, regardless of whether TGF-beta was present in the culture system. SB-431542 abrogated TGF-beta-induced upregulation of alpha-SM-actin, CTGF, and Col I. It effectively inhibited the phosphorylation of Smad2 stimulated by TGF-beta but not that of the components of the MAPK pathways. Conclusions: SB-431542 inhibits scar formation after glaucoma filtration surgery. The mechanism may be that SB-431542 interferes in the phosphorylation of Smad2, thus abrogating TGF-beta-induced fibroblast transdifferentiation and then decreasing Col I synthesis.[3] SB431542 is a potent, selective small-molecule inhibitor of TGF-β superfamily type I ALK receptors (ALK4, ALK5, ALK7) [1] - Its mechanism of action involves competitive binding to the ATP-binding pocket of ALK4/ALK5/ALK7, inhibiting their kinase activity and blocking downstream Smad2/3 phosphorylation and transcriptional activation of TGF-β/activin target genes [1][2][3] - SB431542 exhibits in vitro antiproliferative and anti-fibrotic activities, and in vivo antitumor effects in lung cancer xenografts and anti-scarring effects in ocular filtration surgery models [2][3] - It is widely used as a tool compound to study TGF-β/activin signaling in cancer, fibrosis, development, and tissue repair [1][2][3] - The drug’s selectivity for ALK4/ALK5/ALK7 minimizes off-target effects, supporting its potential therapeutic applications in TGF-β-driven diseases such as cancer and excessive scar formation [2][3] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (5.41 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (5.41 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: 2% DMSO+30% PEG 300+ddH2O: 5mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6015 mL | 13.0076 mL | 26.0152 mL | |
| 5 mM | 0.5203 mL | 2.6015 mL | 5.2030 mL | |
| 10 mM | 0.2602 mL | 1.3008 mL | 2.6015 mL |