Physicochemical Properties
| Molecular Formula | CH7RNPI |
| Molecular Weight | 190.951 |
| Exact Mass | 349.028 |
| CAS # | 1333210-07-3 |
| Related CAS # | SAR7334 hydrochloride;1333207-63-8 |
| PubChem CID | 53378752 |
| Appearance | Light yellow to yellow solid powder |
| Density | 1.5±0.1 g/cm3 |
| Index of Refraction | 1.697 |
| LogP | 2.13 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 4 |
| Rotatable Bond Count | 3 |
| Heavy Atom Count | 26 |
| Complexity | 535 |
| Defined Atom Stereocenter Count | 3 |
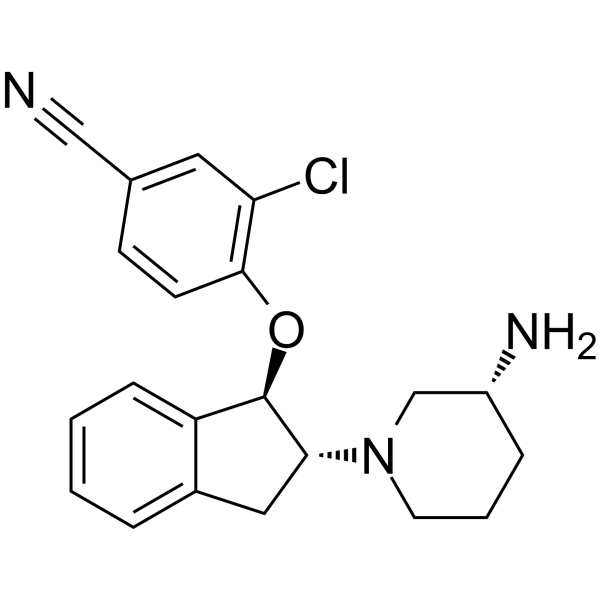
| SMILES | C1C[C@H](CN(C1)[C@@H]2CC3=CC=CC=C3[C@H]2OC4=C(C=C(C=C4)C#N)Cl)N |
| InChi Key | RLKRLNQEXBPQGQ-OZOXKJRCSA-N |
| InChi Code | InChI=1S/C21H22ClN3O/c22-18-10-14(12-23)7-8-20(18)26-21-17-6-2-1-4-15(17)11-19(21)25-9-3-5-16(24)13-25/h1-2,4,6-8,10,16,19,21H,3,5,9,11,13,24H2/t16-,19-,21-/m1/s1 |
| Chemical Name | 4-[[(1R,2R)-2-[(3R)-3-aminopiperidin-1-yl]-2,3-dihydro-1H-inden-1-yl]oxy]-3-chlorobenzonitrile |
| Synonyms | SAR7334; 1333210-07-3; CPA-1588; 4-[[(1R,2R)-2-[(3R)-3-aminopiperidin-1-yl]-2,3-dihydro-1H-inden-1-yl]oxy]-3-chlorobenzonitrile; 4-(((1R,2R)-2-((R)-3-aminopiperidin-1-yl)-2,3-dihydro-1H-inden-1-yl)oxy)-3-chlorobenzonitrile; CHEMBL4129809; SAR-7334; Benzonitrile, 4-[[(1R,2R)-2-[(3R)-3-aMino-1-piperidinyl]-2,3-dihydro-1H-inden-1-yl]oxy]-3-chloro-; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
TRPC6 (IC50 = 7.9 nM) TRPC6 channel (IC₅₀ = 9.5 μM for TRPC6-mediated Ca²⁺ influx) TRPC3 channel (IC₅₀ = 282 μM for TRPC3-mediated Ca²⁺ influx) TRPC7 channel (IC₅₀ = 226 μM for TRPC7-mediated Ca²⁺ influx) Does not affect TRPC4- or TRPC5-mediated Ca²⁺ entry [1] TRPC6 channel (IC₅₀ = 9.5 nM for Ca²⁺ influx; IC₅₀ = 7.9 nM for whole-cell currents) TRPC3 channel (IC₅₀ = 282 nM for Ca²⁺ influx) TRPC7 channel (IC₅₀ = 226 nM for Ca²⁺ influx) Does not affect TRPC4 or TRPC5-mediated Ca²⁺ entry (IC₅₀ > 10 µM). Store-operated Ca²⁺ entry driven by ORAI1/STIM1 is also largely resistant.[3] |
| ln Vitro |
SAR7334 prevents Ca2+ influx into cells mediated by TRPC6, TRPC3, and TRPC7, with IC50 values of 9.5, 282, and 226 nM, respectively[1][2][3]. In contrast, TRPC4- and TRPC5-mediated Ca2+ influx is unaffected. In podocytes, SAR7334 (1 μM) significantly inhibits the calcium influx triggered by Ang II [1]. SAR7334 at 1 μM barely affects SOCE [2]. With an IC50 of 7.9 nM, SAR7334 dose-dependently decreases TRPC6 current. SAR7334 (100 nM) significantly reduces TRPC6 current [3]. SAR7334 (1 μM, 10 min pre-incubation) partially restores glomerular volume change in isolated rat glomeruli attenuated by Ang II, and potently blocks Ang II-evoked calcium influx in podocytes.[1] SAR7334 (1 μM) had negligible effects on the store-operated calcium entry (SOCE) in cultured embryonic (E13) mouse cortical neurons, indicating that TRPC channels (specifically TRPC3, TRPC6, TRPC7) are not critical components of native SOCs in these neurons.[2] SAR7334 potently inhibits diacylglycerol (OAG)-induced Ca²⁺ influx through recombinant human TRPC6 channels expressed in HEK-FITR cells with an IC₅₀ of 9.5 nM. It also inhibits TRPC3 and TRPC7 with lower potency (IC₅₀ = 282 nM and 226 nM, respectively), but has no appreciable effect on TRPC4 or TRPC5 (IC₅₀ > 10 µM). [3] Whole-cell patch-clamp experiments confirm that SAR7334 blocks OAG-activated TRPC6 currents with an IC₅₀ of 7.9 nM. [3] SAR7334 (100 nM) also substantially inhibits TRPC6 currents evoked by receptor stimulation (using trypsin to activate PLC-linked receptors), demonstrating its efficacy against physiologically relevant activation pathways. [3] |
| ln Vivo |
In isolated perfused mouse lungs, SAR7334 (10 mg/kg, orally) suppresses TRPC6-dependent acute HPV. Its appropriateness for long-term oral administration is demonstrated by SAR7334. SAR7334 did not change the mean arterial pressure in spontaneously hypertensive rats (SHR) in an initial short-term trial [3]. In isolated, perfused, and ventilated mouse lungs, SAR7334 dose-dependently suppresses acute hypoxic pulmonary vasoconstriction (HPV), a process exclusively dependent on TRPC6 channels. Half-maximal inhibition was achieved at approximately 100 nM in this ex vivo model. [3] In conscious, spontaneously hypertensive rats (SHR) instrumented with telemetry devices, acute oral administration of SAR7334 (10 mg/kg) did not significantly affect mean arterial blood pressure, suggesting that TRPC6 channels do not play a major role in systemic blood pressure regulation in this model. [3] |
| Enzyme Assay |
Fluo-4 measurement of intracellular calcium concentration ([Ca2+]i)[3] Ca2+ measurements were performed at room temperature using a fluorometric imaging plate reader. Cells grown on black poly-D-lysine-coated 96-well plates were washed with standard extracellular solution (140 mM NaCl, 1 mM MgCl2, 5.4 mM KCl, 2 mM CaCl2, 10 mM HEPES, 10 mM glucose, pH 7.35) and stained with dye solution (2 μM Fluo-4 AM, 0.02% pluronic F127, 0.1% BSA in standard extracellular solution) for 30 min at room temperature. The cells were rinsed and incubated with standard extracellular solution supplemented with different concentrations of the test compound or vehicle for 10 min. Ca2+ entry into TRPC3/6/7-expressing cells was elicited by application of the diacylglycerol, 1-oleoyl-2-acetyl-sn-glycerol (OAG). For calculation of SAR7334-induced inhibition, fluorescence values were plotted over time and the AUC was considered as a measure of Ca2+ influx. |
| Cell Assay |
Measurement of hypoxic pulmonary vasoconstriction (HPV) in isolated perfused and ventilated lungs[3] C57/BL6N mice were anaesthetised with xylazine and ketamine and anticoagulated with heparin as previously described (Weissmann et al., 2004; Fuchs et al., 2011). Male 6–8 weeks old mice were from Charles River Laboratories. In brief, lungs were explanted during deep anaesthesia and artificially ventilated and perfused blood free at 2 mL·min−1 at 37°C with Krebs-Henseleit buffer containing 120 mM NaCl, 4.3 mM KCl, 1.1 mM KH2PO4, 2.4 mM CaCl2, 1.3 mM MgCl2, 13.32 mM glucose, 5% (w/v) hydroxyethylamylopectin and 23.8 mM NaHCO3. The left atrial pressure was set at 2.0 mmHg. Positive pressure ventilation (250 μL tidal volume, 90 breath·min−1 and 2 cm H2O positive end-expiratory pressure) was performed with a mixture containing 21% O2, 5.3% CO2, balanced with N2 (normoxia) or 1% O2, 5.3% CO2 (hypoxia). The pressure in the pulmonary artery and in the left atrium was measured. Changes in pulmonary artery pressure directly reflect changes in pulmonary vascular tone as the lungs are perfused at constant flow. Lungs were ventilated in an alternating mode with hypoxia (10 min) or normoxia (15 min) to induce acute HPV. In such a sequence of repetitive hypoxic ventilation manoeuvres, increasing doses of SAR7334 were applied 5 min prior to the next hypoxic ventilation manoeuvre. For application the SAR7334 stock solution (2 mM in 100% DMSO) was diluted 1:100 in perfusion buffer and appropriate amounts were cumulatively added to the recirculating perfusate (15 mL). The first application was performed after the second hypoxic ventilation period. The strength of acute HPV is given as the maximum increase of pulmonary arterial pressure in each hypoxic ventilation period, referenced to the strength of the second hypoxic manoeuvre (set at 100%). In freshly isolated rat glomeruli, podocytes were pre-incubated with 1 μM SAR7334 for 10 minutes, then challenged with 10 μM Ang II. Calcium influx was monitored using fluorescence imaging (Fluo4/FuraRed ratio). SAR7334 significantly inhibited the Ang II-induced calcium transient.[1] Cortical neurons from embryonic (E13) mice were cultured for 2–3 days in vitro. Cells were loaded with the fluorescent Ca²⁺ probe Fluo-4/AM. To elicit SOCE, cells were maintained in a Ca²⁺-free saline and treated with 200 nM thapsigargin (Tg) to deplete endoplasmic reticulum Ca²⁺ stores. Extracellular Ca²⁺ (2 mM) was then reintroduced to trigger Ca²⁺ influx. SAR7334 (1 μM) was added to the recording saline 60 seconds prior to Tg application and remained present throughout the experiment. Ca²⁺ responses were monitored by time-lapse fluorescence imaging. SAR7334 did not significantly affect the peak amplitude of SOCE in these neurons.[2] For intracellular calcium ([Ca²⁺]i) measurements, recombinant HEK or CHO cells expressing human TRPC3, TRPC6, or TRPC7 channels were used. Cells were seeded on 96-well plates, washed, and loaded with Fluo-4 AM dye. After dye loading, cells were incubated with varying concentrations of SAR7334 or vehicle for 10 minutes. TRPC channel-mediated Ca²⁺ influx was then triggered by applying the diacylglycerol analogue 1-oleoyl-2-acetyl-sn-glycerol (OAG, 30 µM). Fluorescence changes were monitored using a fluorometric imaging plate reader (FLIPR). The area under the fluorescence curve was calculated and used to determine the inhibitory potency (IC₅₀) of SAR7334. [3] For whole-cell patch-clamp electrophysiology, TRPC6-HEK-FITR cells grown on coverslips were used. Cells were continuously perfused with extracellular solution. TRPC6 currents were elicited by either 50 µM OAG or 200 nM trypsin (to mimic receptor activation). SAR7334 was applied cumulatively via the perfusion system. Currents were recorded while holding the cell at -70 mV, and voltage ramps were applied periodically to assess current-voltage relationships. The inhibition of the OAG- or trypsin-induced steady-state current by SAR7334 was quantified to generate dose-response curves. [3] |
| Animal Protocol |
In vivo determination of SAR7334 pharmacokinetics[3] Plasma concentrations of SAR7334 were determined in a serial sampling study after single oral administration of the compound (250 g) in 30% glycopherol/cremophor (75/25) 70% glucose (5%) solution to male Sprague Dawley rats. From each animal, eight plasma samples (approximately 200 µL blood were taken by tail tip sampling) were collected over 24 h and stored below −15°C until analysis. After addition of the precipitant solution (acetonitrile) containing an analogous internal standard, the test item SAR7334 was detected by LC-MS/MS, using an Agilent LC with CTC HTC PAL auto sampler and a Sciex API4000 mass spectrometer in the positive ion mode. Using a sample volume of 50 μL, the lower limit of quantitation was 2.0 ng·mL-1 and the linear range was between 2.0 and 2000 ng·mL−1. Telemetric assessment of BP Adult male (6 months old) spontaneously hypertensive rats were treated on two consecutive days. On day one, the animals received 1 mL·kg-1 vehicle by oral gavage. After 24 h, rats received either vehicle alone or were treated with 10 mg·kg-1 SAR7334. Telemetric measurement of BP was performed as described (Lohn et al., 2009). In brief, a telemetric device was placed between the aorta and the vena cava and the catheter tip of the transmitter was inserted into the aorta. Systolic BP, diastolic BP and heart rate were acquired continuously at a sampling rate of 500 Hz and data were stored as 5 min averages. Mean arterial pressure was calculated from systolic and diastolic pressure and low-pass filtered using the fast Fourier transform function of the vendor software (Dataquest A.R.T. V4.0, Data Sciences International) for better visualization of time-dependent BP variations. For statistical analysis, raw data were averaged over a 6 h period starting 2 h after application of vehicle or SAR7334 (labelled ‘post’). This interval corresponded to the maximal plasma levels of SAR7334 (see Figure 6). Baseline data (labelled ‘pre’) were sampled over the same time interval on the day before treatment. Hypoxic Pulmonary Vasoconstriction (HPV) ex vivo: Lungs were isolated from C57/BL6N mice under anesthesia. They were artificially ventilated and perfused with a blood-free Krebs-Henseleit buffer at constant flow. Acute HPV was induced by alternating ventilation with normoxic (21% O₂) and hypoxic (1% O₂) gas mixtures. SAR7334 was dissolved in DMSO to prepare a 2 mM stock solution. For application, the stock was diluted 1:100 in perfusion buffer and added cumulatively to the recirculating perfusate (15 mL total volume). Increasing doses were applied 5 minutes prior to subsequent hypoxic challenges. The increase in pulmonary artery pressure during hypoxia was measured and normalized to a control response. [3] Pharmacokinetics study: SAR7334 was formulated in a vehicle containing 30% glycopherol/cremophor (75/25) and 70% glucose (5%) solution. Male Sprague Dawley rats received a single oral gavage of SAR7334 at a dose of 10 mg/kg. Serial blood samples (~200 µL) were collected from the tail tip over 24 hours for plasma concentration analysis. [3] Blood pressure telemetry study: Adult male spontaneously hypertensive rats (SHR) were used. A telemetry device was surgically implanted for continuous blood pressure monitoring. In a cross-over design, rats first received vehicle (1 mL/kg) by oral gavage. After 24 hours, the same rats received either vehicle again or SAR7334 (10 mg/kg, p.o.). Blood pressure parameters were recorded continuously. Data from a 6-hour period starting 2 hours post-dose (corresponding to peak plasma levels) were compared to baseline data from the same time interval on the previous day. [3] |
| ADME/Pharmacokinetics |
After a single oral administration of 10 mg/kg to male Sprague Dawley rats, SAR7334 reached pharmacologically effective plasma concentrations that were maintained for several hours. [3] |
| References |
[1]. The Role of Angiotensin II in Glomerular Volume Dynamics and Podocyte Calcium Handling. Sci Rep. 2017 Mar 22;7(1):299. [2]. Pharmacological Characterization of the Native Store-Operated Calcium Channels of Cortical Neurons from Embryonic Mouse Brain. Front Pharmacol. 2016 Dec 12;7:486. [3]. Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels. Br J Pharmacol. 2015 Jul;172(14):3650-60. |
| Additional Infomation |
Podocytes are becoming a primary focus of research efforts due to their association with progressive glomeruli damage in disease states. Loss of podocytes can occur as a result of excessive intracellular calcium influx, and we have previously shown that angiotensin II (Ang II) via canonical transient receptor potential 6 (TRPC6) channels caused increased intracellular Ca2+ flux in podocytes. We showed here with patch-clamp electrophysiology that Ang II activates TRPC channels; then using confocal calcium imaging we demonstrated that Ang II-dependent stimulation of Ca2+ influx in the podocytes is precluded by blocking either AT1 or AT2 receptors (ATRs). Application of Ang(1-7) had no effect on intracellular calcium. Ang II-induced calcium flux was decreased upon inhibition of TRPC channels with SAR7334, SKF 96365, clemizole hydrochloride and La3+, but not ML204. Using a novel 3D whole-glomerulus imaging ex vivo assay, we revealed the involvement of both ATRs in controlling glomerular permeability; additionally, using specific inhibitors and activators of TRPC6, we showed that these channels are implicated in the regulation of glomerular volume dynamics. Therefore, we provide evidence demonstrating the critical role of Ang II/TRPC6 axis in the control of glomeruli function, which is likely important for the development of glomerular diseases.[1] \n\nIn the murine brain, the first post-mitotic cortical neurons formed during embryogenesis express store-operated channels (SOCs) sensitive to Pyr3, initially proposed as a blocker of the transient receptor potential channel of C type 3 (TRPC3 channel). However, Pyr3 does not discriminate between Orai and TRPC3 channels, questioning the contribution of TRPC3 in SOCs. This study was undertaken to clarify the molecular identity and the pharmacological profile of native SOCs from E13 cortical neurons. The mRNA expression of STIM1-2 and Orai1-3 was assessed by quantitative reverse transcription polymerase chain reaction. E13 cortical neurons expressed STIM1-2 mRNAs, with STIM2 being the predominant isoform. Only transcripts of Orai2 were found but no Orai1 and Orai3 mRNAs. Blockers of Orai and TRPC channels (Pyr6, Pyr10, EVP4593, SAR7334, and GSK-7975A) were used to further characterize the endogenous SOCs. Their activity was recorded using the fluorescent Ca2+ probe Fluo-4. Cortical SOCs were sensitive to the Orai blockers Pyr6 and GSK-7975A, as well as to EVP4593, zinc, copper, and gadolinium ions, the latter one being the most potent SOCs blocker tested (IC50 ∼10 nM). SOCs were insensitive to the TRPC channel blockers Pyr10 and SAR7334. In addition, preventing mitochondrial Ca2+ uptake inhibited SOCs which were unaffected by inhibitors of the Ca2+-independent phospholipase A2. Altogether, Orai2 channels are present at the beginning of the embryonic murine cortico-genesis and form the core component of native SOCs in the immature cortex. This Ca2+ route is likely to play a role in the formation of the brain cortex.[2] \n\nBackground and purpose: The cation channel transient receptor potential canonical (TRPC) 6 has been associated with several pathologies including focal segmental glomerulosclerosis, pulmonary hypertension and ischaemia reperfusion-induced lung oedema. We set out to discover novel inhibitors of TRPC6 channels and investigate the therapeutic potential of these agents.\n\nExperimental approach: A library of potential TRPC channel inhibitors was designed and synthesized. Activity of the compounds was assessed by measuring intracellular Ca(2+) levels. The lead compound SAR7334 was further characterized by whole-cell patch-clamp techniques. The effects of SAR7334 on acute hypoxic pulmonary vasoconstriction (HPV) and systemic BP were investigated.\n\nKey results: SAR7334 inhibited TRPC6, TRPC3 and TRPC7-mediated Ca(2+) influx into cells with IC50 s of 9.5, 282 and 226 nM, whereas TRPC4 and TRPC5-mediated Ca(2+) entry was not affected. Patch-clamp experiments confirmed that the compound blocked TRPC6 currents with an IC50 of 7.9 nM. Furthermore, SAR7334 suppressed TRPC6-dependent acute HPV in isolated perfused lungs from mice. Pharmacokinetic studies of SAR7334 demonstrated that the compound was suitable for chronic oral administration. In an initial short-term study, SAR7334 did not change mean arterial pressure in spontaneously hypertensive rats (SHR).\n\nConclusions and implications: Our results confirm the role of TRPC6 channels in hypoxic pulmonary vasoregulation and indicate that these channels are unlikely to play a major role in BP regulation in SHR. SAR7334 is a novel, highly potent and bioavailable inhibitor of TRPC6 channels that opens new opportunities for the investigation of TRPC channel function in vivo.[3] SAR7334 is a novel specific inhibitor of TRPC6 channels, and is used in this study to demonstrate the involvement of TRPC6 in Ang II-mediated glomerular volume dynamics and podocyte calcium handling.[1] SAR7334 was used as a pharmacological tool to investigate the contribution of TRPC channels to native store-operated calcium channels (SOCs) in embryonic cortical neurons. Its lack of effect supported the conclusion that Oral2 channels, not TRPC channels, form the core component of SOCs in this neuronal population.[2] SAR7334 was discovered through a pharmacophore-guided design of focused aminoindanol libraries, based on analogues of the cation channel inhibitor SKF96365. [3] It is a novel, highly potent, and orally bioavailable inhibitor of TRPC6 channels, combining nanomolar activity with a favourable pharmacokinetic profile suitable for chronic studies. [3] Its selectivity profile and in vivo activity make it a valuable pharmacological tool for investigating TRPC6-mediated processes in vivo, with potential therapeutic implications for conditions like focal segmental glomerulosclerosis (FSGS), pulmonary hypertension, and ischemia-reperfusion-induced lung edema. [3] |
Solubility Data
| Solubility (In Vitro) | DMSO : ≥ 370 mg/mL (~1005.79 mM) |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 5.2370 mL | 26.1849 mL | 52.3697 mL | |
| 5 mM | 1.0474 mL | 5.2370 mL | 10.4739 mL | |
| 10 mM | 0.5237 mL | 2.6185 mL | 5.2370 mL |