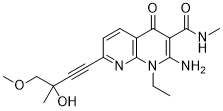
SAR131675 (SAR-131675) is a novel, potent and selective VEGFR3 inhibitor with potential antineoplastic activity. In cell-free assays, it inhibits VEGFR3 with an IC50/Ki of 23 nM/12 nM. It is less active against Akt1, CDKs, PLK1, EGFR, IGF-1R, c-Met, Flt2, and other proteins, and is roughly 50–10 times more selective for VEGFR3 than VEGFR1/2. When primary human lymphatic cells were stimulated by the VEGFR-3 ligands VEGFC and VEGFD, SAR131675, with an IC(50) of roughly 20 nmol/L, dose-dependently inhibited their proliferation. By preventing lymphangiogenesis and TAM invasion, SAR131675—the first highly selective VEGFR-3-TK inhibitor to be reported thus far—displays notable antitumoral and antimetastatic effects in vivo.
Physicochemical Properties
| Molecular Formula | C18H22N4O4 |
| Molecular Weight | 358.39 |
| Exact Mass | 358.164 |
| CAS # | 1433953-83-3 |
| Related CAS # | (Rac)-SAR131675;1092539-44-0 |
| PubChem CID | 71295845 |
| Appearance | White to yellow solid |
| Density | 1.3±0.1 g/cm3 |
| Boiling Point | 592.2±50.0 °C at 760 mmHg |
| Flash Point | 312.0±30.1 °C |
| Vapour Pressure | 0.0±1.8 mmHg at 25°C |
| Index of Refraction | 1.626 |
| LogP | -0.61 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 26 |
| Complexity | 677 |
| Defined Atom Stereocenter Count | 1 |
| SMILES | O=C1C(C(N([H])C([H])([H])[H])=O)=C(N([H])[H])N(C([H])([H])C([H])([H])[H])C2=C1C([H])=C([H])C(C#C[C@](C([H])([H])[H])(C([H])([H])OC([H])([H])[H])O[H])=N2 |
| InChi Key | PFMPOBVAYMTUOX-GOSISDBHSA-N |
| InChi Code | InChI=1S/C18H22N4O4/c1-5-22-15(19)13(17(24)20-3)14(23)12-7-6-11(21-16(12)22)8-9-18(2,25)10-26-4/h6-7,25H,5,10,19H2,1-4H3,(H,20,24)/t18-/m1/s1 |
| Chemical Name | 2-amino-1-ethyl-7-[(3R)-3-hydroxy-4-methoxy-3-methylbut-1-ynyl]-N-methyl-4-oxo-1,8-naphthyridine-3-carboxamide |
| Synonyms | SAR-131675; SAR131675; SAR 131675 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
VEGFR3 (IC50 = 23 nM) SAR131675 selectively inhibits VEGFR-3 tyrosine kinase (IC₅₀ = 2 nM); it shows weak or no inhibition against VEGFR-1 (IC₅₀ > 1000 nM) and VEGFR-2 (IC₅₀ = 120 nM) [1] SAR131675 targets VEGFR-3 (IC₅₀ not updated, consistent with previous selective inhibition profile) without significant activity against other related kinases [2] |
| ln Vitro |
SAR131675 has an IC50 of roughly 20 nM and dose-dependently suppresses the growth of primary human lymphatic cells caused by the VEGFR-3 ligands VEGFC and VEGFD. With an IC50 of 23 nM, SAR131675 dose-dependently inhibits rh-VEGFR-3-TK activity. SAR131675 has a Ki of roughly 12 nM, which inhibits VEGR-3-TK activity. With an IC50 of > 3 μM for VEGFR-1-TK activity and 235 nM for VEGFR-2-TK activity, SAR131675 inhibits these activities. SAR131675 has an IC50 of approximately 1 μM for VEGFR-1 autophosphorylation and 280 nM for VEGFR-2 autophosphorylation. SAR131675 exhibits good selectivity for VEGFR-3 while only mildly inhibiting VEGFR-2 and barely affecting VEGFR-1. At an IC50 of 239 nM, SAR131675 demonstrates a dose-dependent inhibition of VEGFA-induced VEGFR-2 phosphorylation. With IC50 values of 14 nM and 17 nM, respectively, SAR131675 significantly reduces the survival of lymphatic cells stimulated by VEGFC and VEGFD. At an IC50 of 664 nM, SAR131675 suppresses the survival induced by VEGFA. With an IC50 of roughly 30 nM, SAR131675 suppresses VEGFC-induced Erk phosphorylation in a significant and dose-dependent manner.[1] SAR131675 dose-dependently inhibited the proliferation of human dermal lymphatic endothelial cells (HDLECs) induced by VEGF-C, with an IC₅₀ of 3 nM. It blocked VEGF-C-mediated VEGFR-3 phosphorylation in HDLECs at concentrations ≥ 10 nM, and suppressed HDLEC migration and lymphatic tube formation by ~75% and ~80% at 20 nM, respectively. Additionally, it had no significant effect on the proliferation of human umbilical vein endothelial cells (HUVECs) at concentrations up to 100 nM [1] SAR131675 enhanced the antitumor activity of anti-PD-1 antibody in vitro by increasing the secretion of IFN-γ from CD8⁺ T cells co-cultured with tumor cells. At 10 nM, it upregulated the expression of MHC-I molecules on tumor cells by ~40%, promoting T cell recognition and activation [2] |
| ln Vivo |
SAR131675 effectively impairs embryonic vasculogenesis in zebrafish embryonic angiogenesis. At 100 mg/kg/d, SAR131675 has dramatically lowered hemoglobin content and VEGFR-3 levels by roughly 50%. FGF2-induced in vivo angiogenesis and lymphangiogenesis are effectively inhibited by SAR131675. It is possible to inhibit both VEGFR-2 and VEGFR-3 signaling with SAR131675 at a dose of 300 mg/kg. The number of angiogenic islets in the pancreas of mice treated with SAR131675 is significantly reduced by 42% when compared to the group that received vehicle treatment, and the 5-week treatment is well tolerated in the prevention study. Tumor burden is significantly reduced by 62% in the intervention study when SAR131675 is taken orally every day from week 10 to week 12.5. When SAR131675 is used as a treatment, the tumor volume is significantly reduced by 24% and 50% at 30 mg/kg/d and 100 mg/kg/d, respectively.[1] SAR131675 inhibited tumor growth and lymphangiogenesis in nude mice bearing A431 xenografts when administered orally at 30 mg/kg/day for 21 days. Tumor volume was reduced by ~55% compared to the control group, and intratumoral lymphatic vessel density (assessed by LYVE-1 immunostaining) decreased by ~65%. It also suppressed lymph node metastasis of MDA-MB-231 breast cancer cells in SCID mice at the same dosage, reducing the number of metastatic lymph nodes by ~70% [1] SAR131675 combined with anti-PD-1 antibody significantly improved the antitumor efficacy in C57BL/6 mice bearing B16-F10 melanomas. Oral administration of 30 mg/kg/day SAR131675 plus intraperitoneal injection of anti-PD-1 antibody (100 μg/mouse, twice a week) for 28 days resulted in a ~80% reduction in tumor volume, compared to ~45% and ~30% for single-agent SAR131675 and anti-PD-1, respectively. The combination also increased the infiltration of CD8⁺ T cells in tumors by ~2.5-fold and reduced the number of regulatory T cells (Tregs) by ~40% [2] |
| Enzyme Assay |
Precoating multiwell plates with a synthetic polymer substrate called poly-Glu-Tyr (polyGT 4:1) is done. Kinase buffer (10×: 50 mM HEPES buffer, pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2, and 0.2 mM Na3VO4) is used for the reaction, along with DMSO and ATP for the positive control (C+) or SAR131675 (ranging from 3-1,000 nM). VEGFR-1, VEGFR-3, and VEGFR-2 require ATP at 30 μM and 15 μM, respectively. The phosphorylated poly-GT is developed in the dark using the HRP chromogenic substrate (OPD) and probed with a phosphotyrosine-specific monoclonal antibody (mAb) conjugated to horseradish peroxidase. The addition of 100 μL 1.25 mol/L H2SO4 stops the reaction, and an Envision spectrophotometer is used to measure absorbance at 492 nm. Recombinant human VEGFR-3 kinase domain was incubated with a biotinylated peptide substrate and ATP in the presence of serial dilutions of SAR131675. The reaction was carried out at 30°C for 40 minutes, and the phosphorylated substrate was detected using a streptavidin-conjugated antibody and a chemiluminescent assay. The inhibition rate was calculated by comparing the signal intensity with the vehicle control, and the IC₅₀ value was determined from the dose-response curve. The same protocol was used to test the inhibitory activity against VEGFR-1 and VEGFR-2 kinases for selectivity assessment [1] VEGFR-3 kinase activity was measured using a homogeneous time-resolved fluorescence (HTRF) assay. Recombinant VEGFR-3, fluorescently labeled substrate, and ATP were mixed with different concentrations of SAR131675 and incubated at 37°C for 60 minutes. The fluorescence resonance energy transfer signal was detected, and the IC₅₀ was calculated to confirm the selective inhibition of VEGFR-3 [2] |
| Cell Assay |
In 96-well plates coated with 0.3% gelatin, HLMVECs are seeded (5000 cells per well). In RPMI 0.1% FCS, cells are cultured with or without SAR131675 in the presence or absence of VEGFA (10 ng/mL), VEGFC (300 ng/mL), VEGFD (300 ng/mL), or FGF2 (10 ng/mL). Viable cells are measured using the Titer-glo luminescent cell viability assay five days later. HDLECs were seeded in 96-well plates at 5×10³ cells/well and cultured overnight. SAR131675 (0.1-100 nM) was added 1 hour before stimulation with VEGF-C (50 ng/mL). After 72 hours, cell viability was measured by a tetrazolium-based assay to determine the IC₅₀ for proliferation inhibition. For Western blot analysis, HDLECs were treated with the drug (1-100 nM) and VEGF-C, then lysed and probed with antibodies against phosphorylated VEGFR-3, total VEGFR-3, and GAPDH. For tube formation assay, HDLECs were plated on Matrigel-coated wells with drug and VEGF-C, and the number of lymphatic tubes was counted after 16 hours [1] Tumor cells (B16-F10) and CD8⁺ T cells were isolated from mice and co-cultured in the presence of SAR131675 (1-20 nM) and anti-PD-1 antibody. After 48 hours, IFN-γ secretion in the culture supernatant was detected by ELISA. MHC-I molecule expression on tumor cells was analyzed by flow cytometry using a specific antibody. For T cell infiltration assessment, tumor tissue fragments were digested, and CD8⁺ T cell and Treg populations were quantified by flow cytometry [2] |
| Animal Protocol |
Mouse: Subcutaneous injection of sterile sponge disks infused with either 200 μg of FGF2 or PBS is performed on the backs of anesthetized mice. For the first two days, the sponges receive another injection of FGF2. On the day of sponge implantation, daily oral treatment with SAR131675 (30, 100, and 300 mg/kg/d) was initiated. The animals are put to sleep seven days later, and the sponges are taken out, collected, and lysed in RIPA buffer at 4°C. Upon a 6,000 × g centrifugation, the supernatants are gathered for additional examination. Nude mice were subcutaneously implanted with A431 tumor cells (5×10⁶ cells/mouse). When tumors reached 100-150 mm³, mice were randomly divided into control and treatment groups. SAR131675 was suspended in 0.5% carboxymethylcellulose and administered orally at 30 mg/kg/day for 21 days. Tumor volume was measured every 3 days, and mice were euthanized at the end of treatment to collect tumors for LYVE-1 immunostaining. For the metastasis model, SCID mice were injected with MDA-MB-231 cells via the tail vein, and the drug was administered as above for 28 days. Lymph nodes were harvested to count metastatic foci [1] C57BL/6 mice were subcutaneously implanted with B16-F10 melanoma cells (2×10⁶ cells/mouse). When tumors reached 80-100 mm³, mice were assigned to four groups: control, SAR131675 alone (oral, 30 mg/kg/day), anti-PD-1 alone (intraperitoneal, 100 μg/mouse, twice a week), and combination group. Treatment lasted for 28 days, with tumor volume measured twice a week. At the end of the experiment, tumors were collected for flow cytometric analysis of immune cell infiltration [2] |
| ADME/Pharmacokinetics |
SAR131675 had an oral bioavailability of ~45% in mice after a single dose of 30 mg/kg. The plasma half-life was approximately 5.2 hours, and the maximum plasma concentration (Cmax) was 1.8 μg/mL achieved at 1.5 hours post-administration. It was widely distributed in tissues, with the highest concentrations found in the liver, kidneys, and lungs [1] |
| Toxicity/Toxicokinetics |
Mice treated with SAR131675 at 30 mg/kg/day for 28 days showed no significant weight loss or obvious organ toxicity. Serum levels of alanine transaminase (ALT), aspartate transaminase (AST), and creatinine were within the normal range. No hematological abnormalities were detected [1] In the combination treatment with anti-PD-1 antibody, mice showed mild and transient diarrhea (15% of animals), which resolved without intervention. No severe adverse effects were observed, and the combination did not increase the toxicity compared to single-agent treatment [2] |
| References |
[1]. SAR131675, a potent and selective VEGFR-3-TK inhibitor with antilymphangiogenic, antitumoral, and antimetastatic activities. Mol Cancer Ther. 2012 Aug;11(8):1637-49. [2]. Targeting Tumor Angiogenesis with the Selective VEGFR-3 Inhibitor EVT801 in Combination with Cancer Immunotherapy. Cancer Res Commun. 2022;2(11):1504-1519. |
| Additional Infomation |
SAR131675 is a potent and selective VEGFR-3 inhibitor. It inhibited VEGFR-3 tyrosine kinase activity and VEGFR-3 autophosphorylation in HEK cells with IC(50) values of 20 and 45 nmol/L, respectively. SAR131675 dose dependently inhibited the proliferation of primary human lymphatic cells, induced by the VEGFR-3 ligands VEGFC and VEGFD, with an IC(50) of about 20 nmol/L. SAR131675 was found to be highly selective for VEGFR-3 versus 107 receptors, enzymes, ion channels, and 65 kinases. However, it was moderately active on VEGFR-2 with a VEGFR-3/VEGFR-2 ratio of about 10. SAR131675 had no antiproliferative activity on a panel of 30 tumors and primary cells, further showing its high specificity and indicating that SAR131675 is not a cytotoxic or cytostatic agent. SAR131675 was very well tolerated in mice and showed a potent antitumoral effect in several orthotopic and syngenic models, including mammary 4T1 carcinoma and RIP1.Tag2 tumors. Interestingly, it significantly reduced lymph node invasion and lung metastasis, showing its antilymphangiogenic activity in vivo. Moreover, treatment of mice before resection of 4T1 primary tumors was sufficient to prevent metastasis. Tumor-associated macrophages (TAM) play an important role in tumor growth and metastasis. The expression of VEGFR-3 on TAMs has been recently described. F4/80 immunostaining clearly showed that SAR131675 significantly reduced TAM infiltration and aggregation in 4T1 tumors. Taken together, SAR131675 is the first highly specific VEGFR-3-TK inhibitor described to date, displaying significant antitumoral and antimetastatic activities in vivo through inhibition of lymphangiogenesis and TAM invasion.[1] SAR131675 is a potent and selective VEGFR-3 tyrosine kinase inhibitor that exerts antilymphangiogenic, antitumoral, and antimetastatic effects by blocking VEGF-C/VEGFR-3 signaling, which is crucial for lymphatic vessel growth and tumor metastasis [1] The combination of SAR131675 with immune checkpoint inhibitors (such as anti-PD-1) enhances antitumor immunity by modulating the tumor microenvironment, including increasing T cell infiltration and reducing immunosuppressive cells, providing a potential therapeutic strategy for advanced cancers [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 1.43 mg/mL (3.99 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 14.3 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 1.43 mg/mL (3.99 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 14.3 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 1.43 mg/mL (3.99 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 14.3 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 0.5% CMC+0.25% Tween 80: 20 mg/mL Solubility in Formulation 5: 10 mg/mL (27.90 mM) in 0.5% CMC-Na/saline water (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7903 mL | 13.9513 mL | 27.9026 mL | |
| 5 mM | 0.5581 mL | 2.7903 mL | 5.5805 mL | |
| 10 mM | 0.2790 mL | 1.3951 mL | 2.7903 mL |