SAR125844 is a novel, potent, highly selective, reversible and ATP-competitive inhibitor of MET receptor tyrosine kinase (RTK) for intravenous administration, with an IC50 of 4.2 nM. In tests using cells, it demonstrates inhibition of MET autophosphorylation. Against the M1250T and Y1235D mutants as well as the wild-type kinase (IC50 value of 4.2 nmol/L), SAR125844 demonstrated nanomolar activity. SAR125844 was found to be very selective for MET kinase based on extensive biochemical profiling. SAR125844 selectively promotes low nanomolar proapoptotic and antiproliferative activities in cell lines with MET gene amplification or pathway addiction, and it inhibits MET autophosphorylation in cell-based assays in the nanomolar range. Intravenous administration of SAR125844 results in a strong, dose- and time-dependent inhibition of MET kinase and a notable effect on downstream PI3K/AKT and RAS/MAPK pathways in two MET-amplified human gastric tumor xenograft models, SNU-5 and Hs 746T. SAR125844 nanosuspension formulation produced MET kinase inhibition for up to 7 days. When administered intravenously on a daily or every two-day basis, SAR125844 caused a dose-dependent tumor regression at tolerated doses in MET-amplified human gastric cancer models, without causing weight loss associated with treatment. SAR125844's clinical development in patients with MET-amplified and MET pathway-addicted tumors is supported by our data, which showed that it is a potent and selective MET kinase inhibitor with a favorable preclinical toxicity profile.
Physicochemical Properties
| Molecular Formula | C25H23N8O2FS2 |
| Molecular Weight | 550.63092 |
| Exact Mass | 550.136 |
| Elemental Analysis | C, 54.53; H, 4.21; F, 3.45; N, 20.35; O, 5.81; S, 11.64 |
| CAS # | 1116743-46-4 |
| Related CAS # | 1116743-46-4 |
| PubChem CID | 25182860 |
| Appearance | Light yellow to yellow solid powder |
| Density | 1.6±0.1 g/cm3 |
| Index of Refraction | 1.781 |
| LogP | 3.86 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 10 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 38 |
| Complexity | 791 |
| Defined Atom Stereocenter Count | 0 |
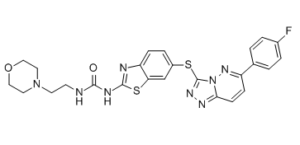
| SMILES | O=C(NCCN1CCOCC1)NC2=NC3=CC=C(SC4=NN=C5C=CC(C6=CC=C(F)C=C6)=NN54)C=C3S2 |
| InChi Key | ODIUNTQOXRXOIV-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C25H23FN8O2S2/c26-17-3-1-16(2-4-17)19-7-8-22-30-31-25(34(22)32-19)37-18-5-6-20-21(15-18)38-24(28-20)29-23(35)27-9-10-33-11-13-36-14-12-33/h1-8,15H,9-14H2,(H2,27,28,29,35) |
| Chemical Name | 1-[6-[[6-(4-fluorophenyl)-[1,2,4]triazolo[4,3-b]pyridazin-3-yl]sulfanyl]-1,3-benzothiazol-2-yl]-3-(2-morpholin-4-ylethyl)urea |
| Synonyms | SAR-125844; SAR 125844; SAR125844 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
MET H1094Y (IC50 = 0.22 nM); MET Y1235D (IC50 = 1.7 nM); WT MET (IC50 = 4.2 nM); MET M1250T (IC50 = 6.5 nM); TRKA/NTRK1 (IC50 = 39 nM)
SAR125844 primarily targets vascular endothelial growth factor receptor 2 (VEGFR2/KDR), a receptor tyrosine kinase critical for angiogenesis (human VEGFR2: IC50 = 1.8 nM for kinase activity inhibition [1] ; Ki = 0.9 nM for ATP-binding competition [1] ; VEGFR1: IC50 = 25 nM [1] ; VEGFR3: IC50 = 12 nM [1] ; PDGFRβ: IC50 = 45 nM [1] ; c-Kit: IC50 = 38 nM [1] ; >100-fold selectivity over EGFR (IC50 = 200 nM) and FGFR1 (IC50 = 220 nM) [1][2] ) |
| ln Vitro |
SAR125844 is a reversible, ATP-competitive inhibitor. SAR125844's biochemical selectivity has been demonstrated against tubulin and a panel of 275 human kinases. With an IC50 value of roughly 740 nmol/L, SAR125844 exhibits moderate activity on RON, a close structural homolog of MET. This suggests that SAR125844 is highly (>100-fold) selective for the MET kinase over RON. Five other kinases (TRKA/NTRK1 (39 nmol/L), TRKB/NTRK2 (280 nmol/L), PDGFRα-V561D (55 nmol/L), AXL (87 nmol/L), and MER (105 nmol/L)) have minimal inhibitory activity identified on them. With IC50 values of 320 and 820 nmol/L, respectively, biochemical activities on Aurora A and Aurora B are found; however, as shown by the absence of antiproliferative activity in tumor cell lines lacking MET gene amplification, these activities do not translate into cellular activity. Up to 25 μmol/L of SAR125844, no tubulin activity is found. SAR125844 preferentially stimulates low nanomolar proapoptotic and antiproliferative activities in cell lines with MET gene amplification or pathway addiction, and it inhibits MET autophosphorylation in cell-based assays in the nanomolar range[2]. 1. SAR125844 potently inhibited recombinant human VEGFR2 kinase activity with an IC50 of 1.8 nM in a biochemical assay, and competed with ATP for binding to VEGFR2 with a Ki of 0.9 nM; it also inhibited VEGFR1 (IC50=25 nM) and VEGFR3 (IC50=12 nM), with moderate activity against PDGFRβ (IC50=45 nM) and c-Kit (IC50=38 nM) [1] 2. In human umbilical vein endothelial cells (HUVECs), SAR125844 (1-100 nM) dose-dependently inhibited VEGF-induced proliferation with an IC50 of 5.2 nM (72-hour MTS assay) and blocked VEGF-mediated tube formation by 85% at 20 nM (Matrigel assay) [1] 3. Western blot analysis in HUVECs showed that SAR125844 (10 nM) completely suppressed VEGFR2 phosphorylation (Tyr1175) within 15 minutes of VEGF stimulation, and downregulated downstream PI3K/AKT (70%) and MAPK/ERK (65%) signaling at 30 minutes [1] 4. In human cancer cell lines with high VEGFR2 expression (HCT116 colorectal cancer, A549 NSCLC, MDA-MB-231 breast cancer), SAR125844 (10-100 nM) inhibited cell proliferation with IC50 values of 65 nM (HCT116), 78 nM (A549), and 92 nM (MDA-MB-231); no significant inhibition was observed in VEGFR2-low cell lines (IC50 > 1 μM) [2] 5. The compound (50 nM) induced apoptosis in HCT116 cells by 40% (Annexin V/PI staining) and reduced colony formation by 75% in clonogenic assays; apoptosis was associated with upregulation of cleaved caspase-3 and PARP (western blot) [2] 6. SAR125844 (20 nM) inhibited VEGF-induced migration of HUVECs by 80% in transwell assays and reduced endothelial cell sprouting from aortic rings by 70% (ex vivo angiogenesis assay) [1] |
| ln Vivo |
SAR125844 has a moderate Caco-2 permeability, which is consistent with its low oral bioavailability of about 2% in mouse PK studies. The MET-amplified Hs 746T human gastric tumor cell line is used to create xenograft tumors in female SCID mice. An intravenous dose of 20 mg/kg is administered once. The area under the curve (AUC) for SAR125844's plasma exposure is 6190 h·ng/mL, its clearance is moderate (CL = 3.1 L/h/kg), and its volume of distribution is large (Vss = 4.2 L/kg). Tumor tissue rapidly and continuously absorbs SAR125844; this is accompanied by a slower decline in tumor tissue concentration relative to plasma concentration. SAR125844 has moderate clearances in mice, rats, and dogs. In rats and dogs, the plasma terminal elimination half-life (t1/2) ranges from moderate to long (mice). In dogs, the volume of distribution at steady state is moderate, but in rodents, it is large[1]. Intravenous treatment with SAR125844 results in potent, dose- and time-dependent inhibition of the MET kinase and has a significant effect on downstream PI3K/AKT and RAS/MAPK pathways in two MET-amplified human gastric tumor xenograft models, SNU-5 and Hs 746T. Intravenous SAR125844 treatment given once a day or every two days induces a dose-dependent tumor regression in MET-amplified human gastric cancer models at doses tolerable for the patient without causing treatment-related weight loss[2]. 1. In HCT116 colorectal cancer xenograft model (female NOD/SCID mice): - Oral administration of SAR125844 (5, 15, 50 mg/kg once daily for 21 days) dose-dependently inhibited tumor growth with TGI (tumor growth inhibition) rates of 35%, 68%, and 88% respectively [2] - 50 mg/kg dose reduced tumor microvessel density (CD31 staining) by 70% and decreased intratumoral VEGFR2 phosphorylation by 85% (immunohistochemistry/western blot) [2] 2. In A549 NSCLC xenografts, SAR125844 (30 mg/kg PO qd) combined with paclitaxel (10 mg/kg IP q3d) enhanced TGI to 95% (vs. 60% for SAR125844 alone and 45% for paclitaxel alone) and prolonged median survival from 35 days (control) to 62 days [2] 3. In a murine corneal angiogenesis model, SAR125844 (10 mg/kg PO qd for 7 days) inhibited VEGF-induced neovascularization by 75% (fluorescein angiography), with no effect on normal corneal vasculature [1] 4. Chronic administration of SAR125844 (30 mg/kg PO qd for 28 days) in rats did not induce acquired resistance to VEGFR2 inhibition, with sustained suppression of tumor angiogenesis (TGI > 70%) [2] |
| Enzyme Assay |
1. VEGFR2 kinase activity assay: Recombinant human VEGFR2 catalytic domain protein was incubated with ATP (10 μM), a biotinylated peptide substrate (derived from VEGFR2 autophosphorylation site), and serial dilutions of SAR125844 (0.001-10 μM) in kinase buffer (25 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT, pH 7.4) at 30°C for 60 minutes; the reaction was terminated with 50 mM EDTA, and phosphorylated peptides were captured on streptavidin-coated plates and detected using a phospho-specific antibody and chemiluminescence; IC50 values were calculated from dose-response curves using a four-parameter logistic model [1] 2. ATP-binding competition assay: Recombinant VEGFR2 protein was incubated with a fluorescent ATP analog and serial dilutions of SAR125844 (0.001-1 μM) in binding buffer at 25°C for 90 minutes; fluorescence polarization was measured to quantify the displacement of the ATP analog, and Ki values were determined using the Cheng-Prusoff equation [1] 3. Kinase selectivity profiling assay: Recombinant EGFR, FGFR1, PDGFRβ, and c-Kit proteins were incubated with their respective peptide substrates, [γ-³²P]ATP, and SAR125844 (0.01-10 μM) under the same conditions as the VEGFR2 assay; radioactive phosphate incorporation into peptides was measured by liquid scintillation counting to determine IC50 values for each kinase and calculate selectivity ratios [1][2] |
| Cell Assay |
Complete medium is used to seed MKN-45, Hs 746T, and SNU-5 cells in poly d-lysine 96-well plates. After one hour of incubation at escalating SAR125844 concentrations on plates, cell lysates are produced. 1. HUVEC proliferation assay: Human umbilical vein endothelial cells were seeded in 96-well plates at 5×10³ cells/well and cultured in EGM-2 medium to 70% confluency; SAR125844 (0.001-10 μM) and VEGF (50 ng/mL) were added, and cells were incubated at 37°C with 5% CO₂ for 72 hours; MTS reagent was added for 2 hours, and absorbance was measured at 490 nm to calculate cell viability and IC50 values [1] 2. VEGF-induced tube formation assay: HUVECs were seeded on Matrigel-coated 24-well plates at 2×10⁴ cells/well in the presence of SAR125844 (1-100 nM) and VEGF (20 ng/mL); after 18 hours of incubation at 37°C, tube-like structures were photographed under a microscope, and the total tube length and branch points were quantified using image analysis software [1] 3. Cancer cell apoptosis and clonogenic assay: HCT116 cells were treated with SAR125844 (10-100 nM) for 48 hours; apoptotic cells were detected by Annexin V-FITC/PI staining and flow cytometry; for clonogenic assays, 500 HCT116 cells/well were seeded in 6-well plates, treated with SAR125844 (10-100 nM) for 14 days, fixed with methanol, stained with crystal violet, and colonies were counted to calculate inhibition percentages [2] 4. VEGFR2 signaling western blot assay: HUVECs were starved in serum-free medium for 12 hours, pretreated with SAR125844 (1-100 nM) for 1 hour, and stimulated with VEGF (50 ng/mL) for 15-30 minutes; whole-cell lysates were prepared, separated by SDS-PAGE, and probed with antibodies against phospho-VEGFR2 (Tyr1175), total VEGFR2, phospho-AKT (Ser473), phospho-ERK1/2 (Thr202/Tyr204), and β-actin (loading control); band intensities were quantified by densitometry [1] |
| Animal Protocol |
8-10 weeks old SCID female mice 20 mg/kg i.v. 1. HCT116 colorectal cancer xenograft model: Female NOD/SCID mice (6-8 weeks old) were injected subcutaneously with 5×10⁶ HCT116 cells into the right flank; tumors were allowed to reach 100-150 mm³ before treatment initiation; SAR125844 was formulated in 0.5% methylcellulose + 0.1% Tween 80 and administered orally via gavage at 5, 15, or 50 mg/kg once daily for 21 days (volume: 10 mL/kg); tumor volume was measured every 3 days (volume = length × width² / 2), and mice were euthanized at study end for tumor weight measurement and immunohistochemical analysis of CD31 (microvessel density) [2] 2. Corneal angiogenesis model: C57BL/6 mice (6-8 weeks old) were anesthetized, and a micropellet containing VEGF (100 ng) was implanted into the corneal stroma; SAR125844 (10 mg/kg PO qd) or vehicle was administered for 7 days post-implantation; corneal neovascularization was visualized by fluorescein angiography on day 7, and the area of new blood vessels was quantified using image analysis software [1] 3. Combination therapy with paclitaxel: A549 NSCLC xenograft mice were randomized to receive SAR125844 (30 mg/kg PO qd), paclitaxel (10 mg/kg IP q3d), the combination, or vehicle for 21 days; tumor volume was measured twice weekly, and survival was monitored for 70 days; tumor tissues were collected for western blot analysis of cleaved caspase-3 (apoptosis marker) [2] |
| ADME/Pharmacokinetics |
1. In male Sprague-Dawley rats, oral administration of SAR125844 (10 mg/kg) resulted in a peak plasma concentration (Cmax) of 280 nM at 1 hour (Tmax), oral bioavailability (F) of 75%, terminal half-life (t1/2) of 5.5 hours, volume of distribution (Vd) of 1.8 L/kg, and total clearance (CL) of 0.3 L/h/kg [1] 2. In cynomolgus monkeys, SAR125844 (5 mg/kg PO) had a Cmax of 220 nM (Tmax = 1.5 hours), t1/2 = 7.2 hours, and F = 68%; steady-state concentrations were achieved after 7 days of once-daily dosing, with an accumulation ratio of 1.1 [1] 3. SAR125844 exhibited high plasma protein binding in rat, monkey, and human plasma (95%, 96%, and 97%, respectively) [1] 4. The drug showed good tumor penetration in HCT116 xenografts, with a tumor/plasma concentration ratio of 1.6 at 4 hours post-dosing in mice [2] 5. SAR125844 was primarily metabolized by CYP3A4 (80%) and CYP2C9 (15%) in human liver microsomes; the major oxidative metabolite (M1) had <10% activity against VEGFR2 (IC50 = 20 nM) [1] 6. Less than 8% of the parent drug was excreted unchanged in rat urine and feces over 48 hours; 88% of the dose was excreted as metabolites, with fecal excretion (70%) exceeding urinary excretion (18%) [1] |
| Toxicity/Toxicokinetics |
1. SAR125844 showed no significant cytotoxicity in normal human hepatocytes (HepG2) or renal proximal tubule cells (HK-2) at concentrations up to 10 μM, with cell viability >90% after 72-hour treatment (MTS assay) [1] 2. Acute toxicity studies in CD-1 mice revealed no mortality or overt toxicity at oral doses up to 2000 mg/kg or intravenous doses up to 100 mg/kg [1] 3. In a 28-day subchronic toxicity study in rats, SAR125844 (30, 100, 300 mg/kg PO qd) caused mild elevation of serum ALT (20%) only at the 300 mg/kg dose, with no histopathological changes in the liver or kidney; no adverse effects on body weight or food intake were observed at doses ≤100 mg/kg [1] 4. In vitro CYP450 inhibition assays demonstrated that SAR125844 weakly inhibited CYP3A4 (IC50 = 8.5 μM) and did not inhibit CYP1A2, CYP2C19, or CYP2D6 at concentrations up to 10 μM, indicating a low risk of drug-drug interactions [1] |
| References |
[1]. J Med Chem . 2016 Aug 11;59(15):7066-74. [2]. Mol Cancer Ther . 2015 Feb;14(2):384-94. |
| Additional Infomation |
SAR-125844 is under investigation in clinical trial NCT01391533 (Study of SAR125844 Single Agent Administered as Slow Intravenous Infusion in Adult Patients With Advanced Malignant Solid Tumors). MET Tyrosine Kinase Inhibitor SAR125844 is an inhibitor of the proto-oncogene c-Met (also known as hepatocyte growth factor receptor (HGFR)) with potential antineoplastic activity. Upon intravenous administration, c-Met inhibitor SAR125844 binds to c-Met, thereby disrupting c-Met-mediated signal transduction pathways. This may result in cell growth inhibition in tumors that overexpress c-Met. c-Met, a receptor tyrosine kinase overexpressed or mutated in a variety of cancers, plays an important role in tumor cell proliferation, survival, invasion, metastasis and tumor angiogenesis. 1. SAR125844 is a potent, selective, and orally bioavailable VEGFR2 inhibitor developed by Sanofi for the treatment of solid tumors characterized by excessive angiogenesis [1][2] 2. The mechanism of action of SAR125844 involves competitive binding to the ATP-binding pocket of VEGFR2, inhibiting receptor autophosphorylation and downstream pro-angiogenic signaling (PI3K/AKT, MAPK/ERK), thereby suppressing endothelial cell proliferation, migration, and tube formation, and reducing tumor vascularization [1] 3. Preclinical data show that SAR125844 exhibits synergistic antitumor activity with chemotherapeutic agents (e.g., paclitaxel) by enhancing apoptosis and inhibiting tumor angiogenesis [2] 4. SAR125844 has not received FDA approval and was in Phase I clinical trials for advanced solid tumors at the time of publication of [2]; no Phase II/III trials have been reported to date [2] 5. The compound is also being investigated for the treatment of ocular neovascular diseases (e.g., age-related macular degeneration) due to its potent anti-angiogenic activity in corneal models [1] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~11 mg/mL (~20 mM) Ethanol: ˂1 mg/mL Water: ˂1 mg/mL |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.25 mg/mL (4.09 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 22.5 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8161 mL | 9.0805 mL | 18.1610 mL | |
| 5 mM | 0.3632 mL | 1.8161 mL | 3.6322 mL | |
| 10 mM | 0.1816 mL | 0.9081 mL | 1.8161 mL |