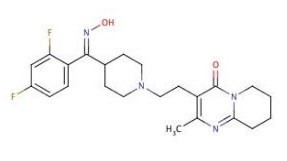
Risperidone E-oxime is an impurity of Risperidone (R-64766; Risperdal), which is an marketed atypical antipsychotic and a potent multi-targeted antagonist for dopamine, serotonin, adrenergic and histamine receptors.
Physicochemical Properties
| Molecular Formula | C₂₃H₂₈F₂N₄O₂ |
| Molecular Weight | 430.49 |
| Exact Mass | 430.218 |
| CAS # | 691007-09-7 |
| PubChem CID | 10048326 |
| Appearance | Typically exists as solid at room temperature |
| Density | 1.3±0.1 g/cm3 |
| Boiling Point | 569.0±60.0 °C at 760 mmHg |
| Flash Point | 297.9±32.9 °C |
| Vapour Pressure | 0.0±1.6 mmHg at 25°C |
| Index of Refraction | 1.631 |
| LogP | 3.16 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 31 |
| Complexity | 774 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | Cc1c(c(=O)n2c(n1)CCCC2)CCN3CCC(CC3)/C(=N\O)/c4ccc(cc4F)F |
| InChi Key | BRCINVRBDDVLDW-QYQHSDTDSA-N |
| InChi Code | InChI=1S/C23H28F2N4O2/c1-15-18(23(30)29-10-3-2-4-21(29)26-15)9-13-28-11-7-16(8-12-28)22(27-31)19-6-5-17(24)14-20(19)25/h5-6,14,16,31H,2-4,7-13H2,1H3/b27-22- |
| Chemical Name | 3-[2-[4-[(Z)-C-(2,4-difluorophenyl)-N-hydroxycarbonimidoyl]piperidin-1-yl]ethyl]-2-methyl-6,7,8,9-tetrahydropyrido[1,2-a]pyrimidin-4-one |
| Synonyms | Risperidone Eoxime; Risperidone E-Oxime; 691007-09-7; 2-Fluoro risperidone E-oxime; UNII-MG83X8A10T; MG83X8A10T; Risperidone specified impurity A [EP]; 4H-Pyrido(1,2-a)pyrimidin-4-one, 3-(2-(4-((E)-(2,4-difluorophenyl)(hydroxyimino)methyl)-1-piperidinyl)ethyl)-6,7,8,9-tetrahydro-2-methyl-; RISPERIDONE IMPURITY A [EP IMPURITY]; Risperidone E oxime |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Risperidone impurity |
| References |
[1]. 5-HT2 and D2 dopamine receptor occupancy in the living human brain. A PET study with risperidone. Psychopharmacology (Berl). 1993;110(3):265-72. [2]. Risperidone and paliperidone inhibit p-glycoprotein activity in vitro. Neuropsychopharmacology. 2007 Apr;32(4):757-64. |
| Additional Infomation |
It has been suggested that a combined blockade of 5-HT2 and D2 dopamine receptors may be superior to D2 dopamine antagonists alone in the treatment of schizophrenia. Risperidone, which has a high affinity for 5-HT2 and D2 dopamine receptors in vitro, is a new antipsychotic drug that has been developed according to this hypothesis. The aim of this study was to examine if risperidone indeed induces 5-HT2 and D2 dopamine receptor occupancy in vivo in humans. Central receptor occupancy was examined by positron emission tomography (PET) in three healthy men after oral administration of 1 mg risperidone. [11C]N-methylspiperone ([11C]NMSP) was used as a radioligand for determination of 5-HT2 receptor occupancy in the neocortex. Both an equilibrium ratio analysis and a kinetic three-compartmental analysis indicated a 5-HT2 receptor occupancy about 60%. [11C]raclopride was used as a radioligand for determination of D2 dopamine receptor occupancy in the striatum and the calculated occupancy was about 50%. This is the first quantitative determination of 5-HT2 receptor occupancy induced by an antipsychotic drug in the living human brain. The results indicate that 5-HT2 receptor occupancy should be very high at the dose level of 4-10 mg risperidone daily, as suggested for clinical use. Risperidone is thus an appropriate compound for clinical evaluation of the benefit of combined 5-HT2 and D2 dopamine receptor blockade in the treatment of schizophrenia. [1] Risperidone (RSP) and its major active metabolite, 9-hydroxy-risperidone (paliperidone, PALI), are substrates of the drug transporter P-glycoprotein (P-gp). The goal of this study was to examine the in vitro effects of RSP and PALI on P-gp-mediated transport. The intracellular accumulation of rhodamine123 (Rh123) and doxorubicin (DOX) were examined in LLC-PK1/MDR1 cells to evaluate P-gp inhibition by RSP and PALI. Both compounds significantly increased the intracellular accumulation of Rh123 and DOX in a concentration-dependent manner. The IC(50) values of RSP for inhibiting P-gp-mediated transport of Rh123 and DOX were 63.26 and 15.78 microM, respectively, whereas the IC(50) values of PALI were >100 microM, indicating that PALI is a less potent P-gp inhibitor. Caco-2 and primary cultured rat brain microvessel endothelial cells (RBMECs) were utilized to investigate the possible influence of RSP on intestinal absorption and blood-brain barrier (BBB) transport of coadministered drugs that are P-gp substrates. RSP, 1-50 microM, significantly enhanced the intracellular accumulation of Rh123 in Caco-2 cells by inhibiting P-gp activity with an IC(50) value of 5.87 microM. Following exposure to 10 microM RSP, the apparent permeability coefficient of Rh123 across Caco-2 and RBMECs monolayers was increased to 2.02 and 2.63-fold in the apical to basolateral direction, but decreased to 0.37 and 0.21-fold in the basolateral to apical direction, respectively. These data suggest that RSP and PALI, to a lesser extent, have a potential to influence the pharmacokinetics and hence the pharmacodynamics of coadministered drugs via inhibition of P-gp-mediated transport. However, no human data exist that address this issue. In particular, RSP may interact with its own active metabolite PALI by promoting its brain concentration through inhibiting P-gp-mediated efflux of PALI across endothelial cells of the BBB. [2] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3229 mL | 11.6147 mL | 23.2293 mL | |
| 5 mM | 0.4646 mL | 2.3229 mL | 4.6459 mL | |
| 10 mM | 0.2323 mL | 1.1615 mL | 2.3229 mL |