Raxatrigine (also known as GSK1014802 and CNV-1014802) is a novel, potent small molecule state-dependent sodium channel blocker, the Nav1.7 sodium channel inhibitor. It has the potential to be used as an analgesic and is being developed by Convergence Pharmaceuticals for the treatment of lumbosacral radiculopathy (sciatica) and trigeminal neuralgia (TGN).
Physicochemical Properties
| Molecular Formula | C18H19FN2O2 |
| Molecular Weight | 314.3541 |
| Exact Mass | 314.143 |
| Elemental Analysis | C, 68.77; H, 6.09; F, 6.04; N, 8.91; O, 10.18 |
| CAS # | 934240-30-9 |
| Related CAS # | Raxatrigine hydrochloride;934240-31-0; 934240-30-9; 934240-35-4 (mesylate) |
| PubChem CID | 16046068 |
| Appearance | White to off-white solid powder |
| LogP | 4.161 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 4 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 23 |
| Complexity | 399 |
| Defined Atom Stereocenter Count | 2 |
| SMILES | C1C[C@H](N[C@H]1C2=CC=C(C=C2)OCC3=CC=CC=C3F)C(=O)N |
| InChi Key | JESCETIFNOFKEU-SJORKVTESA-N |
| InChi Code | InChI=1S/C18H19FN2O2/c19-15-4-2-1-3-13(15)11-23-14-7-5-12(6-8-14)16-9-10-17(21-16)18(20)22/h1-8,16-17,21H,9-11H2,(H2,20,22)/t16-,17+/m1/s1 |
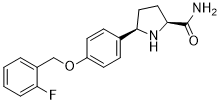
| Chemical Name | (2S,5R)-5-(4-((2-fluorobenzyl)oxy)phenyl)pyrrolidine-2-carboxamide |
| Synonyms | CNV1014802; CNV-1014802; CNV 1014802; 934240-30-9; Vixotrigine; GSK1014802; BIIB074; GSK-1014802; GSK1014802; GSK 1014802; GSK-1014802; Raxatrigine. |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Sodium channel Nav1.7
Voltage-gated sodium channels (Nav) subtypes (Nav1.7: IC50 = 3.2 μM for peak current inhibition; Nav1.2: IC50 = 5.8 μM; Nav1.3: IC50 = 4.5 μM; Nav1.8: IC50 = 7.1 μM) [2] Voltage-gated sodium channels (non-selective subtype inhibition) [1] |
| ln Vitro |
GSK2 and GSK3 have the same ability to stop PCP-induced reversal learning deficits as lamotrigine, indicating that they may be used to treat cognitive symptoms of schizophrenia. Nonetheless, greater dosages than those needed for the medication's anticonvulsant efficacy are necessary for the reversal learning model to be active, indicating a narrow therapeutic window in comparison to the mechanism-dependent central adverse effects of this indication. The US Food and medicine Administration designated Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) as an orphan medicine in July 2013. The Nav1.7 channel represents a promising target for pain relief. In the recent decades, a number of Nav1.7 channel inhibitors have been developed. According to the effects on channel kinetics, these inhibitors could be divided into two major classes: reducing activation or enhancing inactivation. To date, however, only several inhibitors have moved forward into phase 2 clinical trials and most of them display a less than ideal analgesic efficacy, thus intensifying the controversy regarding if an ideal candidate should preferentially affect the activation or inactivation state. In the present study, we investigated the action mechanisms of a recently clinically confirmed inhibitor Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) using both electrophysiology and site-directed mutagenesis. We found that CNV1014802 inhibited Nav1.7 channels through stabilizing a nonconductive inactivated state. When the cells expressing Nav1.7 channels were hold at 70 mV or 120 mV, the half maximal inhibitory concentration (IC50) values (with 95% confidence limits) were 1.77 (1.20-2.33) and 71.66 (46.85-96.48) μmol/L, respectively. This drug caused dramatic hyperpolarizing shift of channel inactivation but did not affect activation. Moreover, CNV1014802 accelerated the onset of inactivation and delayed the recovery from inactivation. Notably, application of CNV1014802 (30 μmol/L) could rescue the Nav1.7 mutations expressed in CHO cells that cause paroxysmal extreme pain disorder (PEPD), thereby restoring the impaired inactivation to those of the wild-type channel. Our study demonstrates that CNV1014802 enhances the inactivation but does not reduce the activation of Nav1.7 channels, suggesting that identifying inhibitors that preferentially affect inactivation is a promising approach for developing drugs targeting Nav1.7 [2]. Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) exerts a state-dependent inhibition on Nav1.7 channels. [2] Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) does not affect steady-state activation. [2] Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) causes a hyperpolarizing shift in the steady-state inactivation. [2] Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) shows use-dependent inhibition of Nav1.7 channels. [2] Influences of Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) on the development of inactivation. [2] Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) slows the recovery from inactivation. [2] Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) normalizes the functional effects of PEPD mutations. [2] 1. Nav channel modulation (Nav1.7-focused): Raxatrigine (also known as CNV1014802/Vixotrigine) acts as a Nav channel modulator that preferentially enhances channel inactivation rather than reducing activation. In HEK293 cells stably expressing human Nav1.7, the compound dose-dependently inhibited peak sodium currents with an IC50 of 3.2 μM. It shifted the steady-state inactivation curve of Nav1.7 to more negative potentials (-58 mV vs. -52 mV in vehicle control) and prolonged the recovery from inactivation (τ = 12.8 ms vs. 5.3 ms in vehicle control at 10 μM). No significant effect on channel activation kinetics was observed [2] 2. Broad Nav subtype inhibition: In cultured rat cortical neurons, Raxatrigine (1–30 μM) inhibited tetrodotoxin-sensitive (TTX-S) and tetrodotoxin-resistant (TTX-R) sodium currents, with IC50 values of 6.4 μM and 8.7 μM, respectively. This inhibition reduced PCP-induced abnormal neuronal hyperexcitability, as reflected by a 45% decrease in spontaneous action potential frequency at 10 μM [1] 3. Metabolic stability: In human liver microsomes, Raxatrigine showed moderate metabolic stability with a half-life of 2.8 hours. It was primarily metabolized by CYP3A4, with no major active metabolites detected [3] |
| ln Vivo |
Sodium channel inhibition is a well precedented mechanism used to treat epilepsy and other hyperexcitability disorders. The established sodium channel blocker and broad-spectrum anticonvulsant lamotrigine is also effective in the treatment of bipolar disorder and has been evaluated in patients with schizophrenia. Double-blind placebo-controlled clinical trials found that the drug has potential to reduce cognitive symptoms of the disorder. However, because of compound-related side-effects and the need for dose titration, a conclusive evaluation of the drug's efficacy in patients with schizophrenia has not been possible. (5R)-5-(4-{[(2-Fluorophenyl)methyl]oxy}phenyl)-l-prolinamide (GSK2) and (2R,5R)-2-(4-{[(2-fluorophenyl)methyl]oxy}phenyl)-7-methyl-1,7-diazaspiro[4.4]nonan-6-one (GSK3) are two new structurally diverse sodium channel blockers with potent anticonvulsant activity. In this series of studies in the rat, we compared the efficacy of the two new molecules to prevent a cognitive deficit induced by the N-methyl-d-aspartic acid receptor antagonist phencyclidine (PCP) in the reversal-learning paradigm in the rat. We also explored the effects of the drugs to prevent brain activation and neurochemical effects of PCP. We found that, like lamotrigine, both GSK2 and GSK3 were able to prevent the deficit in reversal learning produced by PCP, thus confirming their potential in the treatment of cognitive symptoms of schizophrenia. However, higher doses than those required for anticonvulsant efficacy of the drugs were needed for activity in the reversal-learning model, suggesting a lower therapeutic window relative to mechanism-dependent central side effects for this indication. [1] 1. Prevention of PCP-induced cognitive dysfunction in rats: Male Sprague-Dawley rats were intraperitoneally injected with phencyclidine (PCP, 5 mg/kg) once daily for 7 days to induce cognitive impairment. Raxatrigine was administered intraperitoneally at doses of 3 mg/kg, 10 mg/kg, or 30 mg/kg 30 minutes before PCP injection. In the Morris water maze test, the 10 mg/kg and 30 mg/kg groups showed significantly reduced escape latency (45 ± 8 s and 38 ± 6 s vs. 72 ± 10 s in PCP control) and increased time spent in the target quadrant (35 ± 5% and 42 ± 4% vs. 22 ± 3% in PCP control). In the passive avoidance test, these doses also prolonged step-through latency (180 ± 20 s and 220 ± 15 s vs. 85 ± 12 s in PCP control), indicating improved learning and memory [1] 2. Analgesic efficacy in mouse models: In the mouse formalin test, intraperitoneal administration of Raxatrigine (10 mg/kg, 30 mg/kg) dose-dependently reduced licking/biting behavior in both the acute (0–10 min: 35 ± 5 s and 20 ± 3 s vs. 85 ± 8 s in control) and inflammatory (11–60 min: 65 ± 7 s and 38 ± 5 s vs. 150 ± 12 s in control) phases. In the hot plate test, the 30 mg/kg dose significantly prolonged paw withdrawal latency (25 ± 3 s vs. 10 ± 2 s in control) at 60 minutes post-administration [2] 3. Safety and tolerability in healthy humans: Single oral doses of Raxatrigine (10–160 mg) and repeat doses (40 mg twice daily for 14 days) were well-tolerated in healthy volunteers. No dose-limiting toxicities were reported. Mild to moderate adverse events included headache (18%), dizziness (12%), and nausea (8%), which resolved spontaneously. No significant changes in vital signs, ECG parameters (including QTc interval), or laboratory values (liver/kidney function, hematology) were observed [3] |
| Enzyme Assay |
Electrophysiology [2] Whole-cell patch-clamp recordings were conducted at room temperature using an Axopatch 200B patch clamp amplifier. Pipettes were pulled from borosilicate glass capillaries with an electrode resistance typically ranging from 1.5 to 4 MΩ. The recording pipette intracellular solution contained the following (in mmol/L): 140 CsF, 10 NaCl, 10 HEPES, 1.1 EGTA and 20 glucose (pH 7.3 adjusted by CsOH); the bath or extracellular solution contained the following (in mmol/L): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES and 20 glucose (pH 7.3 adjusted by NaOH). During the recording, the bath solution was continuously perfused using a BPS perfusion system. Recording was performed after a 5-min equilibration period at −80 mV after breaking into the whole-cell configuration. Currents were acquired at a 50-kHz sampling frequency and filtered at 2 kHz. Series resistance compensation was used and set to 80%. P/N subtraction was never applied throughout the experiment. An unsaturated IC90 concentration (10 μmol/L) was applied throughout the whole study unless otherwise stated. If a change in activation was not observed at 10 μmol/L, a higher concentration of the drug (30 μmol/L) was further administered to confirm the lack of an effect. This higher concentration was also used in the parallel experiments, such as the inactivation recordings. 1. Nav1.7 channel current recording (patch-clamp assay): HEK293 cells stably transfected with human Nav1.7 cDNA were seeded on glass coverslips and cultured for 24–48 hours. Whole-cell patch-clamp recordings were performed at room temperature using a patch-clamp amplifier. The intracellular solution contained potassium aspartate, KCl, MgATP, and EGTA, while the extracellular solution contained NaCl, KCl, CaCl2, and glucose. Raxatrigine was dissolved in extracellular solution at gradient concentrations (0.1–30 μM) and applied to cells via perfusion. Sodium currents were elicited by depolarizing steps from a holding potential of -120 mV to various test potentials (-60 mV to +40 mV). Current-voltage relationships, steady-state inactivation curves, and recovery from inactivation were analyzed to quantify the drug's effect on Nav1.7 channel gating. IC50 values were calculated from concentration-response curves of peak current inhibition [2] 2. Neuronal sodium current assay: Rat cortical neurons were isolated and cultured for 7–10 days. TTX-S and TTX-R sodium currents were recorded using the whole-cell patch-clamp technique. Raxatrigine was applied at concentrations of 1–30 μM, and current amplitude was measured before and after drug application. The percentage of current inhibition was calculated, and IC50 values were determined for both TTX-S and TTX-R currents [1] |
| Cell Assay |
Cell culture and transfection [2] Human embryonic kidney 293 (HEK293) cells stably expressing hNav1.7 were used. Cells were grown in high-glucose DMEM supplemented with 10% fetal bovine serum and were selected with 300 μg/mL of the antibiotic Hygromycin B under standard tissue culture conditions (5% CO2, 37 °C). For the functional expression of Nav1.7 mutants, Chinese hamster ovary (CHO) cells were used and cultured in 50/50 DMEM/F-12 supplemented with 10% fetal bovine serum. Two days prior to recording, the constructs were transfected into CHO cells with Lipofectamine reagent, according to the manufacture's protocol. A GFP construct was co-transfected to aid in the identification of transfected cells by fluorescence microscopy. Cells were seeded onto poly-L-lysine-coated glass coverslips before they were used for electrophysiology recording. 1. Nav1.7-expressing HEK293 cell culture and viability assay: HEK293 cells transfected with Nav1.7 were cultured in DMEM medium supplemented with serum and antibiotics. Cells were seeded in 96-well plates at 5×10³ cells/well and treated with Raxatrigine (0.1–100 μM) for 24 hours. Cell viability was measured using a colorimetric assay, and the CC50 value (>100 μM) was determined, indicating no significant cytotoxicity [2] 2. Cortical neuron culture and hyperexcitability assay: Rat embryonic cortical neurons were dissociated and plated on poly-D-lysine-coated coverslips. After 7 days in culture, neurons were treated with PCP (10 μM) alone or in combination with Raxatrigine (1–30 μM) for 24 hours. Spontaneous action potentials were recorded using patch-clamp electrophysiology, and the frequency and amplitude of action potentials were analyzed to assess the drug's ability to reverse PCP-induced hyperexcitability [1] 3. Human liver microsomal metabolic stability assay: Human liver microsomes were incubated with Raxatrigine (1 μM) in a reaction mixture containing NADPH-regenerating system (glucose-6-phosphate, glucose-6-phosphate dehydrogenase, NADP⁺, MgCl2) at 37°C. Samples were collected at 0, 15, 30, 60, and 120 minutes, and the reaction was terminated by adding acetonitrile. The concentration of Raxatrigine was quantified by LC-MS/MS, and the in vitro half-life was calculated [3] |
| Animal Protocol |
Single ascending dose study procedures[3] \nThis was a double‐blind crossover study conducted at a single clinical site from May 2007 to May 2008. Volunteers and all site personnel were blinded to study treatment allocation but sponsor personnel were unblinded to assist with appropriate dose selection decisions. Eligible volunteers were healthy men aged 18–65 years or healthy women with no childbearing potential aged 18–50 years. Volunteers were also required to be nonsmokers and have a body weight of > 50 kg and body mass index of 19–29.9 kg/m2 (± 10%). Exclusion criteria included significant abnormalities found on clinical examination, or clinical chemistry or hematology parameters. The sample size of 10 participants per cohort is a commonly used number in early studies.\n[3] \nThe steps recommended in the US Food and Drug Administration’s Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers were followed for the estimation of the starting dose. On normalizing the experimentally determined nontoxic dosage level for surface area, the most sensitive preclinical animal species examined was the dog. Using the conversion factor provided in the guidance, the nontoxic dosage level of 70 mg/kg/day in the dog translates to a human equivalent dose of 2,333 mg/day for a 60 kg human. Dividing this value by a conservatively estimated safety factor of 10, the maximum recommended starting dose of Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) was determined to be 233 mg/day; however, the results observed using Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) in pain models indicate that a pharmacologically active dosage of Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) is 1 mg/kg. This efficacious dose is expected to translate to a predicted clinical dose of 10 mg/day in a 60 kg human; thus, a starting dose of 10 mg q.d. was selected.\n[3] \nVolunteers were recruited into 3 cohorts of 10 and treated with a starting dose of Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) 10 mg or placebo in cohort 1 (Figure S1 ). In each dosing session, 8 volunteers received vixotrigine and 2 volunteers received placebo, except cohort 2, dosing session 3 (vixotrigine, n = 4; placebo, n = 6). The highest vixotrigine dose tested in 1 cohort was the initial dose in the subsequent cohort (Figure S2 ). Vixotrigine doses were escalated up to 825 mg, until predefined safety or PK stopping limits were reached. Plasma samples were taken at baseline and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, 48, and 72 hours after dosing. Each volunteer received a maximum of 4 vixotrigine doses and 1 placebo dose over 5 dosing sessions, with the exception of cohort 2 (2 vixotrigine doses and 1 placebo dose over 3 dosing sessions). Each session was separated by a ≥ 7‐day washout period. Volunteers attended a follow‐up visit ~ 7–14 days following the last dose of study medication.\n[3] \nThe primary endpoints of the SAD study were to (i) evaluate Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) safety and tolerability assessed through adverse events (AEs), vital signs (blood pressure, heart rate, and respiration rate), clinical laboratory evaluations (hematology, clinical chemistry, and urinalysis), and 12‐lead electrocardiograms (ECGs); and (ii) evaluate the following vixotrigine PK parameters: area under the concentration‐time curve from time 0 (predose) extrapolated to infinite time (AUC0–inf), AUC from predose to last time of quantifiable concentration (AUC0–t), maximum observed concentration (Cmax), and time to Cmax (Tmax). Dose proportionality of AUC0–inf, AUC0–t, and Cmax across doses was investigated by a power model fitted by restricted maximum likelihood method, with log(dose) fitted as covariate. The intercept for volunteers was fitted as a random effect. Estimated mean slope (β) and 90% confidence intervals were constructed for each parameter.\n[3] \n\nMultiple ascending dose study procedures[3] \nThis single‐blind study included 4 cohorts of parallel staggered doses. Eligible volunteers were healthy men or healthy women with no childbearing potential aged 18–55 years, and a body weight ≥ 50 kg and body mass index ≥ 19 kg/m2 and ≤ 29 kg/m2. No significant abnormalities on clinical examination or through evaluation of clinical chemistry or hematology parameters were permitted.\n[3] \nRaxatrigine (GSK1014802; Vixotrigine; CNV-1014802) was supplied as 50, 100, or 200 mg film‐coated brownish yellow tablets. Placebo tablets visually matched the active tablets and all tablets were taken with 240 mL of water. Twelve volunteers in each of 4 parallel‐dose cohorts were randomized to vixotrigine or placebo in a 9:3 ratio. For all cohorts, a screening phase preceded study treatment and a follow‐up visit was conducted 7–14 days after the last dose. Cohort 1 received one 14‐day repeat‐dose phase (vixotrigine 150 mg q.d. or placebo). Cohort 2 received one 14‐day repeat‐dose phase (vixotrigine 400 mg q.d. or placebo). An additional dose of study drug was administered on day 15, within 30 minutes of consuming a high‐fat breakfast. Cohort 3 received an SD and 28‐day repeat‐dose of vixotrigine 300–400 mg b.i.d. (doses individually adjusted to keep the AUC below the originally defined PK limits) or placebo, with a morning dose given for the SD and on day 28 of the repeat‐dose period. Cohort 4 received an SD and 14‐day repeat‐dose of vixotrigine 350–450 mg b.i.d. (doses individually adjusted to keep below the PK limits for Cmax and AUC) or placebo, with a morning dose given for the SD and on day 15 of the repeat‐dose period. In addition, an assessment of exploratory endpoints (mechanical pain threshold, and pressure pain threshold and tolerance) was completed after day 1 and day 15 SD (see Supplementary Material Table S1 and Figure S3 ). Volunteers in cohorts 3 and 4 received the same treatment allocation (vixotrigine or placebo) in the SD and repeat‐dose periods, which were separated by ≥ 7 days. Predose blood samples were drawn and at prespecified time points to measure plasma vixotrigine levels (0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 hours; cohorts 1 and 4: days 1, 7, and 14; cohort 2: days 1, 7, 14, and 15; cohort 3: days 1, 7, 14, and 28).\n[3] \nThe primary endpoints of the repeat‐dose study were (i) to evaluate the safety and tolerability of Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) by monitoring AEs and concomitant medication, 12‐lead ECGs, lead II monitoring, 24‐hour Holter monitoring, vital signs, and laboratory parameters; and (ii) PK parameters estimated from plasma concentration‐time profiles for each analyte: Cmax, Tmax, and AUC from time 0 (predose) to 24 hours after dosing (AUC0–24; q.d. dose), AUC from time 0 (predose) to 12 hours after dosing (b.i.d. dose), and terminal half‐life (t 1/2).\n[3] \nFor both studies, plasma concentrations of Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) were determined using liquid chromatography‐tandem mass spectrometry after protein precipitation extraction, according to validated analytical methods at LGC. 17 The lower limit of quantification for vixotrigine was 10 ng/mL. PK parameters were derived using noncompartmental analyses with WinNonlin software version 5.0.1. Statistical analyses used SAS version 9.1. 1. PCP-induced cognitive dysfunction rat model: Male Sprague-Dawley rats (200–250 g) were randomly divided into 5 groups (n=8 per group): normal control, PCP control, Raxatrigine 3 mg/kg, 10 mg/kg, and 30 mg/kg. Raxatrigine was dissolved in 0.9% normal saline with 5% DMSO (final DMSO concentration ≤5%) and administered via intraperitoneal injection 30 minutes before PCP (5 mg/kg, intraperitoneal) once daily for 7 days. The normal control group received vehicle instead of PCP and Raxatrigine. On days 8–12, the Morris water maze test was performed (5-day training phase, 1-day probe test). On day 13, the passive avoidance test was conducted (training session: rats were placed in the bright compartment and allowed to enter the dark compartment, followed by a mild foot shock; test session: 24 hours later, step-through latency was recorded) [1] 2. Mouse analgesic models: - Formalin test: Male ICR mice (20–25 g) were randomly divided into 4 groups (n=10 per group): vehicle control, Raxatrigine 10 mg/kg, 30 mg/kg, and positive control. Raxatrigine was dissolved in saline and administered intraperitoneally 30 minutes before intraplantar injection of 5% formalin (20 μL) into the right hind paw. Licking/biting time of the injected paw was recorded in two phases (0–10 min and 11–60 min) [2] - Hot plate test: Mice were placed on a hot plate maintained at 55 ± 0.5°C, and the paw withdrawal latency was recorded before (baseline) and 30, 60, 90 minutes after intraperitoneal administration of Raxatrigine (30 mg/kg) or vehicle. A cut-off time of 30 seconds was set to avoid tissue damage [2] 3. Healthy volunteer clinical study: This was a phase I, randomized, double-blind, placebo-controlled study in healthy adults (18–45 years old). Participants were assigned to single-dose cohorts (10, 20, 40, 80, 160 mg Raxatrigine or placebo) or repeat-dose cohort (40 mg Raxatrigine twice daily for 14 days or placebo). Raxatrigine was administered as oral tablets. Blood samples were collected at predefined time points for pharmacokinetic analysis. Safety assessments included adverse event monitoring, vital signs, 12-lead ECG, and laboratory tests (hematology, chemistry, urinalysis) [3] |
| ADME/Pharmacokinetics |
Single ascending dose PK [3] No quantifiable vixotrigine concentrations were reported in predose plasma samples, indicating no carryover between dosing periods. Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) was rapidly and extensively absorbed, with Cmax generally achieved at 1–2 hours postdose. Dose proportionality was approximate (Figure S4); although statistical significance was not confirmed for vixotrigine 10–825 mg, AUC0–inf showed no relevant deviation from dose proportionality (estimate of the slope from the power model, 1.088). The deviation from dose proportionality for Cmax was larger but still of limited importance (slope, 1.202). Following the maximal vixotrigine dose for this study (825 mg), Cmax and AUC0–inf were 6.53 μg/mL and 66.2 μg*h/mL, respectively (Figure 1). The estimated values for oral clearance and volume of distribution were 13.8 L/hr and 262 L, respectively. The concentration of vixotrigine increased with dose (Figure 2) and there were no dose‐dependent changes in total clearance of vixotrigine from plasma or volume of distribution, indicating linear kinetics. Vixotrigine appeared to have moderate plasma clearance and tissue distribution, with a t 1/2 of ~ 11 hours (Table 1). Multiple ascending dose PK [3] Repeat‐dose PK parameters for Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) are summarized in Table 2. PK characteristics of single oral doses of vixotrigine 150–400 mg were in alignment with those reported in the SD study. Tmax was achieved in ~ 2 hours postdose and t 1/2 was 9–13 hours. Accumulation was observed following repeat vixotrigine doses; dose‐proportional increases in exposure, as measured by AUC0–24 and Cmax, were approximate (Figure S6). As expected, accumulation was higher after b.i.d. dosing by approximately twofold compared with q.d. dosing (Figure 3). Steady‐state of vixotrigine was generally achieved for all repeat‐dose regimens from day 5 onward. When administered a high‐fat meal, vixotrigine 400 mg q.d. AUC0–24 decreased by 3%, Cmax decreased by 15%, and Tmax was delayed by an average of 2.5 hours (Figure S5). Similar to SD PK, no dose‐dependent changes in oral clearance and volume of distribution were observed following repeat‐dose administration of vixotrigine. 1. Absorption: After single oral administration of Raxatrigine (10–160 mg) to healthy volunteers, peak plasma concentrations (Cmax) were reached at a median Tmax of 1.5–2.5 hours. Cmax and AUC0-∞ increased approximately proportionally with dose over the tested range, indicating linear pharmacokinetics. Oral bioavailability was estimated to be ~32% based on comparison with intravenous data (from a separate substudy) [3] 2. Distribution: The apparent volume of distribution (Vd/F) was 18–22 L, suggesting moderate tissue distribution. Plasma protein binding was ~90% (determined by equilibrium dialysis in human plasma), with no concentration-dependent binding (0.1–10 μg/mL) [3] 3. Metabolism: Raxatrigine was primarily metabolized by cytochrome P450 3A4 (CYP3A4) in the liver. The major metabolite was an inactive N-dealkylated product, accounting for ~60% of circulating metabolites. No active metabolites contributing to pharmacologic effects were identified [3] 4. Excretion: The mean plasma elimination half-life (t1/2) was 6.8 ± 1.2 hours after single doses and 7.5 ± 1.5 hours after repeat doses. Approximately 70% of the administered dose was excreted in feces (primarily as metabolites) and 25% in urine (10% as unchanged drug, 15% as metabolites) within 72 hours [3] 5. Clearance: Apparent oral clearance (CL/F) was 1.8–2.2 L/hour, and renal clearance was 0.35 L/hour [3] |
| Toxicity/Toxicokinetics |
Safety and tolerability [3] Single ascending dose safety and tolerability [3] In the SD study, Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) doses up to 825 mg were well‐tolerated in healthy volunteers (Table 3). Twenty‐three (77%) volunteers reported at least 1 AE. Dizziness was the most commonly reported AE (n = 11; 37%), with a higher incidence at higher vixotrigine doses (600 and 825 mg, reported by 4 of 10 (40%) and 5 of 7 (71%) volunteers, respectively). No other AEs appeared to increase with dose. Drug‐related AEs were reported by 14 (47%) volunteers (Table 3); dizziness was again the most commonly reported AE (n = 9; 30%). [3] The majority of AEs following Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) administration were mild in nature, except for 8 events (4 for dizziness and 1 for each of somnolence, headache, diarrhea, and vasovagal syncope associated with sinus node pause) that were rated as moderate. No deaths, serious AEs, withdrawals due to an AE, drug‐related serious AEs, or clinically significant changes in ECG values or clinical laboratory evaluations were reported. Multiple ascending dose safety and tolerability [3] Repeat Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) doses were well‐tolerated at all dose levels up to 450 mg in healthy volunteers (Table 4). An AE was reported by 11 (92%) placebo‐treated volunteers and by 9 (75%), 6 (67%), 9 (100%), and 7 (78%) volunteers treated with vixotrigine 150 mg q.d., 400 mg q.d., 300–400 mg b.i.d., and 350–450 mg b.i.d., respectively. Headache was the most commonly reported AE, with a similar incidence in vixotrigine‐treated and placebo‐treated volunteers. Any drug‐related AE was reported by 6 (50%), 3 (25%), 4 (44%), 8 (89%), and 6 (67%) volunteers across the placebo and vixotrigine 150 mg q.d., 400 mg q.d., 300–400 mg b.i.d., and 350–450 mg b.i.d. treatment groups, respectively. Dizziness was the most frequent drug‐related AE, reported by 1 (8%), 0, 2 (22%), 3 (33%), and 3 (33%) of the placebo and vixotrigine 150 mg q.d., 400 mg q.d., 300–400 mg b.i.d., and 350–450 mg b.i.d. treatment groups, respectively. [3] All AEs were mild in nature, with the exception of 2 volunteers with vomiting of moderate intensity following placebo, and 2 volunteers with vomiting of moderate intensity following Raxatrigine (GSK1014802; Vixotrigine; CNV-1014802) (1 for each of the 400 mg q.d. and 300–400 mg b.i.d. groups) treatment. No deaths, severe AEs, serious drug‐related AEs, clinically significant abnormalities in ECG values or clinical laboratory evaluations, or withdrawals due to an AE were reported in the repeat‐dose study. 1. In vitro cytotoxicity: Raxatrigine showed no significant cytotoxicity in Nav1.7-expressing HEK293 cells or rat cortical neurons at concentrations up to 100 μM (cell viability >90%) [1][2] 2. Acute in vivo toxicity: Intraperitoneal administration of Raxatrigine at doses up to 300 mg/kg in rats and mice caused no mortality or severe clinical signs. Mild transient sedation was observed at doses ≥100 mg/kg, which resolved within 4 hours [1][2] 3. Subchronic toxicity: Repeat oral administration of Raxatrigine (40 mg/kg twice daily for 14 days) in rats resulted in no significant changes in body weight, organ weights, or histopathological findings in major organs (liver, kidney, heart, brain) [3] 4. Clinical safety: In healthy volunteers, Raxatrigine did not cause clinically significant changes in liver function (ALT, AST, bilirubin), renal function (creatinine, eGFR), or hematology parameters (hemoglobin, white blood cell count). No QTc interval prolongation (ΔQTcF < 10 ms) was observed at any dose [3] 5. Drug-drug interaction potential: In vitro studies showed that Raxatrigine did not inhibit or induce major CYP enzymes (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4) at therapeutic concentrations, indicating low potential for drug-drug interactions [3] |
| References |
[1]. The efficacy of sodium channel blockers to prevent phencyclidine-induced cognitive dysfunction in the rat: potential for novel treatments for schizophrenia. J Pharmacol Exp Ther. 2011 Jul;338(1):100-13. [2]. Enhancing inactivation rather than reducing activation of Nav1.7 channels by a clinically effective analgesic CNV1014802. Acta Pharmacol Sin . 2018 Apr;39(4):587-596. [3]. Safety, Tolerability and Pharmacokinetics of Single and Repeat Doses of Vixotrigine in Healthy Volunteers. Clin Transl Sci. 2020 Dec 13;14(4):1272–1279. |
| Additional Infomation |
Vixotrigine has been investigated for the treatment of Bipolar Disorder and Bipolar Depression. Neuropathic pain affects ~ 6.9–10% of the general population and leads to loss of function, anxiety, depression, sleep disturbance, and impaired cognition. Here, we report the safety, tolerability, and pharmacokinetics of a voltage‐dependent and use‐dependent sodium channel blocker, vixotrigine, currently under investigation for the treatment of neuropathic pain conditions. The randomized, placebo‐controlled, phase I clinical trials were split into single ascending dose (SAD) and multiple ascending dose (MAD) studies. Healthy volunteers received oral vixotrigine as either single doses followed by a ≥ 7‐day washout period for up to 5 dosing sessions (SAD, n = 30), or repeat doses (once or twice daily) for 14 and 28 days (MAD, n = 51). Adverse events (AEs), maximum observed vixotrigine plasma concentration (Cmax), area under the concentration‐time curve from predose to 24 hours postdose (AUC0–24), time to Cmax (Tmax), and terminal half‐life (t 1/2), among others, were assessed. Drug‐related AEs were reported in 47% and 53% of volunteers in the SAD and MAD studies, respectively, with dizziness as the most commonly reported drug‐related AE. SAD results showed that Cmax and AUC increased with dose, Tmax was 1–2 hours, and t 1/2 was ~ 11 hours. A twofold increase in accumulation was observed when vixotrigine was taken twice vs. once daily (MAD). Steady‐state was achieved from day 5 onward. These data indicate that oral vixotrigine is well‐tolerated when administered as single doses up to 825 mg and multiple doses up to 450 mg twice daily.[3] The Nav1.7 channel represents a promising target for pain relief. In the recent decades, a number of Nav1.7 channel inhibitors have been developed. According to the effects on channel kinetics, these inhibitors could be divided into two major classes: reducing activation or enhancing inactivation. To date, however, only several inhibitors have moved forward into phase 2 clinical trials and most of them display a less than ideal analgesic efficacy, thus intensifying the controversy regarding if an ideal candidate should preferentially affect the activation or inactivation state. In the present study, we investigated the action mechanisms of a recently clinically confirmed inhibitor CNV1014802 using both electrophysiology and site-directed mutagenesis. We found that CNV1014802 inhibited Nav1.7 channels through stabilizing a nonconductive inactivated state. When the cells expressing Nav1.7 channels were hold at 70 mV or 120 mV, the half maximal inhibitory concentration (IC50) values (with 95% confidence limits) were 1.77 (1.20-2.33) and 71.66 (46.85-96.48) μmol/L, respectively. This drug caused dramatic hyperpolarizing shift of channel inactivation but did not affect activation. Moreover, CNV1014802 accelerated the onset of inactivation and delayed the recovery from inactivation. Notably, application of CNV1014802 (30 μmol/L) could rescue the Nav1.7 mutations expressed in CHO cells that cause paroxysmal extreme pain disorder (PEPD), thereby restoring the impaired inactivation to those of the wild-type channel. Our study demonstrates that CNV1014802 enhances the inactivation but does not reduce the activation of Nav1.7 channels, suggesting that identifying inhibitors that preferentially affect inactivation is a promising approach for developing drugs targeting Nav1.7.[2] 1. Drug aliases and classification: Raxatrigine is also known by the研发 codes CNV1014802 and Vixotrigine. It is a non-selective voltage-gated sodium channel modulator with preferential activity on Nav1.7, developed for the treatment of neuropathic pain and cognitive dysfunction associated with schizophrenia [1][2][3] 2. Mechanism of action: Unlike traditional sodium channel blockers that reduce channel activation, Raxatrigine enhances the inactivation of Nav channels (especially Nav1.7), which reduces abnormal neuronal hyperexcitability without compromising normal neuronal function. This unique mechanism contributes to its analgesic efficacy and cognitive-improving effects [2] 3. Therapeutic potential: The drug has shown efficacy in preclinical models of neuropathic pain and schizophrenia-related cognitive impairment. Phase I clinical studies confirm its favorable safety and tolerability profile, supporting further development for these indications [1][3] 4. Clinical development status: Raxatrigine has completed Phase I and Phase II clinical trials for neuropathic pain (e.g., post-herpetic neuralgia) and is being evaluated for other indications such as trigeminal neuralgia and cognitive dysfunction [3] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~83 mg/mL (~264.04 mM) |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.1812 mL | 15.9058 mL | 31.8117 mL | |
| 5 mM | 0.6362 mL | 3.1812 mL | 6.3623 mL | |
| 10 mM | 0.3181 mL | 1.5906 mL | 3.1812 mL |