Physicochemical Properties
| Molecular Formula | C29H34FN3O3 |
| Molecular Weight | 491.60 |
| Exact Mass | 491.26 |
| Elemental Analysis | C, 70.85; H, 6.97; F, 3.86; N, 8.55; O, 9.76 |
| CAS # | 3034664-39-3 |
| Appearance | Typically exists as solids at room temperature |
| LogP | 4.8 |
| Hydrogen Bond Donor Count | 0 |
| Hydrogen Bond Acceptor Count | 3 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 25 |
| Complexity | 471 |
| Defined Atom Stereocenter Count | 1 |
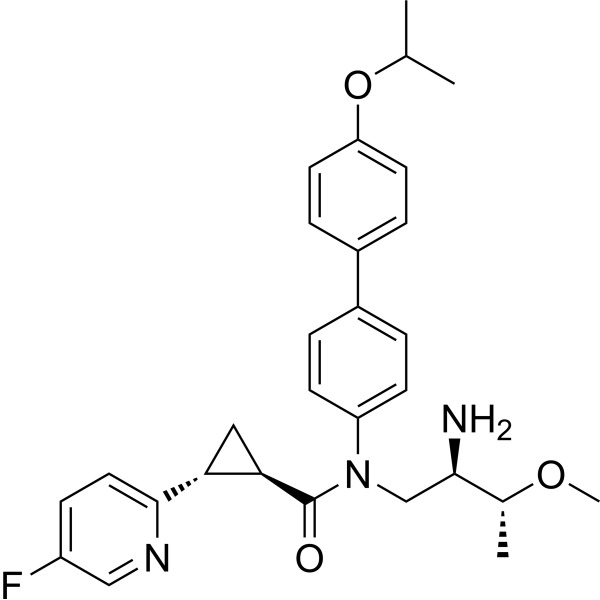
| SMILES | CC(C)OC1=CC=C(C2=CC=C(N(C([C@H]3[C@H](C4=CC=C(F)C=N4)C3)=O)C[C@H]([C@@H](C)OC)N)C=C2)C=C1 |
| InChi Key | OXODNABCLPQQDT-CEWWTIOHSA-N |
| InChi Code | InChI=1S/C29H34FN3O3/c1-18(2)36-24-12-7-21(8-13-24)20-5-10-23(11-6-20)33(17-27(31)19(3)35-4)29(34)26-15-25(26)28-14-9-22(30)16-32-28/h5-14,16,18-19,25-27H,15,17,31H2,1-4H3/t19-,25-,26-,27-/m1/s1 |
| Chemical Name | (1R,2R)-N-((2R,3R)-2-amino-3-methoxybutyl)-2-(5-fluoropyridin-2-yl)-N-(4'-isopropoxy-[1,1'-biphenyl]-4-yl)cyclopropane-1-carboxamide InChi Key: OXODNABCLPQQDT-CEWWTIOHSA-N |
| Synonyms | Rti 122; 3-(4-Iodophenyl)tropan-2beta-carboxylic acid phenyl ester; 146145-22-4; phenyl (1R)-3-(4-iodophenyl)-8-methyl-8-azabicyclo[3.2.1]octane-2-carboxylate; 8-Azabicyclo(3.2.1)octane-2-carboxylic acid, 3-(4-iodophenyl)-8-methyl-, phenyl ester, (1R-(exo,exo))-; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | GPR88 |
| ln Vitro | RTI-122 (30a) has EC50 values of 11 and 12 nM in the cell-based cAMP accumulation and [35S]GTPγS binding assays, respectively, and has GPR88-specific agonist activity in mouse striatal tissues [1]. |
| ln Vivo |
RTI-122 has a favorable PK profile exhibiting good metabolic stability (half-life = 5.8 h) and brain permeability (brain:plasma ratio = 1.2) for in vivo assessment. Finally, RTI-122 at a dose of 10 mg/kg significantly attenuates binge-like alcohol drinking in mice when administrated intraperitoneally. Taken together, RTI-122 is an advanced lead compound for further development of GPR88 agonists for treating AUD and possibly other disease conditions mediated by GPR88. Future studies of RTI-122 include assessments of its oral bioavailability, off-target activity (e.g., hERG), toxicity, and behavioral effects in different alcohol drinking and seeking models [1].
RTI-122 (30a) Reduced Alcohol Drinking.[1] Both genetic and pharmacological studies have suggested that GPR88 is a promising drug target for the treatment of alcohol addiction.9,11,13 We previously demonstrated that mice pre-treated with RTI-13591–33 (30 mg/kg, i.p.), the first brain-penetrant GPR88 agonist, were less likely to drink and seek alcohol.13 Thus, we examined whether 30a would achieve the same effects in vivo. As shown in Figure 3, RTI-13951–33 at a dose of 30 mg/kg significantly reduced alcohol intake [via one-way ANOVA, F(2, 42) = 4.30 and p = 0.02] in the drinking-inthe-dark (DID) procedure. Notably, pretreatment of 30a at a dose of 10 mg/kg produced the same degree of attenuation of binge-like alcohol consumption as that of RTI-13951–33 at a dose of 30 mg/kg (in the Newman–Keuls post hoc test, p < 0.05 compared to saline-treated mice). No effect on water intake was detected (data not shown). The better in vivo potency of 30a than of RTI-13951–33 is likely due to the improved potency and PK properties (metabolic stability and brain penetration) of 30a compared to those of RTI-13951–33. |
| Enzyme Assay |
Kinetic Solubility Assay. [1] The kinetic solubility of RTI-122 (30a) was measured in phosphate-buffered saline (PBS) at pH 7.4. Ten microliters of a test compound stock solution (20 mM DMSO) was combined with 490 μL of PBS (1 mM potassium phosphate monobasic, 3 mM sodium phosphate dibasic, and 155 mM sodium chloride buffer) to reach a targeted concentration of 400 μM. The solution was agitated on a VX-2500 multitube vortexer (VWR) for 2 h at room temperature. Following agitation, the samples were filtered on a glass-fiber filter (1 μm), and the eluates were diluted 400-fold with a 1:1 acetonitrile/water mixture. On each experimental occasion, nicardipine and imipramine were assessed as reference compounds for low and high solubility, respectively. All samples were assessed in triplicate and analyzed by LC-MS/MS using electrospray ionization against standards prepared in the same matrix. Plasma–Protein Binding Assay. [1] The plasma–protein binding of RTI-122 (30a) was determined in C57BL/6 mouse plasma using the high-throughput dialysis (HTD) device. Test compound and controls (1 μM) were spiked in Wistar rat plasma and aliquoted in triplicate in a HTD 96-well plate, where the plasma and dialysate buffer were separated by a semipermeable cellulose membrane. Once sealed, the HTD plate was incubated at 37 °C and kept under light agitation for 6 h, until equilibrium was reached. Plasma and buffer samples were then extracted along with their corresponding standard curve samples using an ice-cold 1:1 acetonitrile/methanol mixture. After centrifugation, supernatants from both plasma- and buffer-containing samples were further diluted prior to being submitted to bioanalysis by LC-MS/MS. Acebutolol and warfarin served as weakly and strongly bound controls, respectively. Liver Microsomal Stability Assay. [1] The liver microsomal stability assessment was performed by Paraza Pharma Inc. Compounds were incubated with mouse liver microsomes at 37 °C for a total of 45 min. The reaction was performed at pH 7.4 in 100 mM potassium phosphate buffer containing 0.5 mg/mL mouse liver microsomal protein. Phase I metabolism was assessed by adding NADPH to a final concentration of 1 mM and collecting samples at 0, 5, 15, 30, and 45 min. All collected samples were quenched with ice-cold stop solution (1 μM labetalol and 1 μM glyburide in acetonitrile) and centrifuged to remove precipitated protein. Resulting supernatants were further diluted with H2O. Diphenhydramine (t1/2 = 9.3 min, and CL = 148 μL min−1 mg−1) and verapamil (t1/2 = 2.2 min, and CL = 630 μL min−1 mg−1) were used as reference compounds. Samples were analyzed by LC-MS/MS, and calculations for half-life and in vitro clearance were accomplished using Microsoft Excel (see the Supporting Information for calculation methods and examples of crude data). |
| Cell Assay |
Bidirectional MDCK-MDR1 Permeability Assay. [1] MDCK-MDR1 cells at passage 12 were seeded onto permeable polycarbonate supports in 12-well Costar Transwell plates and allowed to grow and differentiate for 3 days. On day 3, culture medium (DMEM supplemented with 10% FBS) was removed from both sides of the transwell inserts, and cells were rinsed with warm HBSS. After the rinse step, the chambers were filled with warm transport buffer [HBSS containing 10 mM HEPES and 0.25% BSA (pH 7.4)], and the plates were incubated at 37 °C for 30 min prior to transepithelial electric resistance (TEER) measurements. The buffer in the donor chamber (apical side for the A-to-B assay, basolateral side for the B-to-A assay) was removed and replaced with the working solution (10 μM test article in transport buffer). The plates were then placed at 37 °C under light agitation. At designated time points (30, 60, and 90 min), an aliquot of transport buffer from the receiver chamber was removed and replenished with fresh transport buffer. Reactions were quenched with ice-cold acetonitrile containing an internal standard and then centrifuged to pellet protein. The resulting supernatants were further diluted with a 50:50 acetonitrile/water mixture (water only for atenolol) and subjected to LC-MS/MS analysis. The reported apparent permeability (Papp) values were calculated from a single determination. Atenolol and propranolol were tested as low- and moderate-permeability references, respectively. Bidirectional transport of digoxin was assessed to demonstrate Pgp activity and/or expression. The apparent permeability (Papp, measured in centimeters per second) of a compound was determined according to the formula Papp = (dQ/dt)/(ACi × 60), where dQ/dt is the net rate of appearance in the receiver compartment, A is the area of the Transwell measured in square centimeters (1.12 cm2), Ci is the initial concentration of the compound added to the donor chamber, and 60 is the factor for converting minutes to seconds. |
| Animal Protocol |
Pharmacokinetic Analysis. [1] Male C57BL/6NCrl mice were procured from Charles River Laboratories at 8–9 weeks of age. Animals were allowed to acclimate to the vivarium for a period of 1 week. Each mouse was administered a single 10 mg/kg dose of RTI-122 (30a) through i.p. injection in a 10 mL/kg dosing volume. Doses were formulated in saline. Three animals were sacrificed at each of the selected time points (0.25, 0.5, 1, 2, 4, 6, 8, and 24 h) postdose. Blood was collected through the abdominal vena cava for plasma analysis, and whole-body perfusion was performed with phosphate-buffered saline (pH 7.4). Brains were collected following perfusion. Drinking-in-the-Dark Experiment. [1] Male C57BL/6J mice were individually housed under a 12 h reversed light/dark cycle. Oral alcohol intake was determined using the DID paradigm for 2 weeks. The procedure was adapted from ref 28. Briefly, each week, drinking sessions were conducted 2 h every day during three consecutive days and 4 h on the fourth and final day, with one bottle containing tap water and the other containing alcohol diluted to 20% alcohol (v/v) in tap water. The bottles were weighed every day, and the mice were weighed at the beginning of the experiment. Animals were injected with saline, RTI-13951–33 (30 mg/kg, i.p.), or RTI-122 (30a) (10 mg/kg, i.p.) 1 h before being tested in the 4 h drinking session. Drugs. [1] The alcohol solution was prepared from absolute ethanol (190 proof) diluted to 20% alcohol (v/v). RTI-13951–33 (30 mg/kg) or RTI-122 (30a) (10 mg/kg) was dissolved in sterile 0.9% saline to be administered i.p. at 10 mL/kg. |
| ADME/Pharmacokinetics |
In Vitro ADME and PK Profile of 30a.
RTI-122 (30a) was assessed in a panel of ADME assays, including MDCK-MDR1 permeability, MLM stability, kinetic solubility, and plasma–protein binding (PPB) assays. As shown in Table 4, 30a demonstrated good BBB permeability in the MDCK-MDR1 bidirectional transport assay with an apparent permeability Papp (A → B) of 2.8 × 10–6 cm/s and an efflux ratio (PB→A/PA→B) of 2.5. Notably, 30a had a half-life of 24.6 min and a CL of 56.4 μL min−1 mg−1 in MLMs, which are 10 times better than those of RTI-13951–33 (half-life = 2.2 min, and CL = 643 μL min−1 mg−1). Compound 30a had good aqueous solubility (kinetic solubility = 261 μM) but a high level of protein binding of 99.5% in mouse plasma, which needs to be further optimized. The PK properties of RTI-122 (30a) were evaluated in mice using a single i.p. injection of 10 mg/kg, and the results are summarized in Table 5. Following injection, 30a had good plasma exposure with a Cmax of 662 ng/mL and an AUC0–inf of 7292 ng·h/mL. In line with the MLM data, 30a had good metabolic stability with a half-life of 5.8 h and a CL of 23 mL min−1 kg−1 in plasma. The brain concentration peaked at 4 h with a Cmax of 630 ng/mL (1.3 μM). The overall brain:plasma AUC ratio (Kp) is 1.2, indicating good brain penetration. On the basis of the in vitro ADME and PK findings, compound 30a was selected for in vivo efficacy evaluation.[1] |
| References |
[1]. Improvement of the Metabolic Stability of GPR88 Agonist RTI-13951-33: Design, Synthesis, and Biological Evaluation. J Med Chem. 2023 Feb 23;66(4):2964-2978. |
| Additional Infomation | The orphan receptor GPR88 has recently attracted considerable attention as a novel drug target for the treatment of neuropsychiatric disorders, including Parkinson’s disease, schizophrenia, and drug addiction. To date, the endogenous ligand for GPR88 has not been discovered. To characterize GPR88 signaling mechanisms and biological functions, our laboratory, as well as others, has carried out a medicinal chemistry campaign to develop GPR88 synthetic agonists. RTI-13951–33 is the first-generation in vivo active agonist that has been shown to reduce alcohol drinking and seeking behaviors in mice and rats. In the study presented here, we have conducted a lead optimization to further improve the potency and PK profile of RTI-13951–33. Our medicinal chemistry efforts led to the identification of RTI-122 (30a) that has EC50 values of 11 and 12 nM in the cell-based cAMP accumulation and [35S]GTPγS binding assays, respectively, and has GPR88-specific agonist activity in mouse striatal tissues. In addition, RTI-122 has a favorable PK profile exhibiting good metabolic stability (half-life = 5.8 h) and brain permeability (brain:plasma ratio = 1.2) for in vivo assessment. Finally, RTI-122 at a dose of 10 mg/kg significantly attenuates binge-like alcohol drinking in mice when administrated intraperitoneally. Taken together, RTI-122 is an advanced lead compound for further development of GPR88 agonists for treating AUD and possibly other disease conditions mediated by GPR88. Future studies of RTI-122 include assessments of its oral bioavailability, off-target activity (e.g., hERG), toxicity, and behavioral effects in different alcohol drinking and seeking models. [1] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0342 mL | 10.1709 mL | 20.3417 mL | |
| 5 mM | 0.4068 mL | 2.0342 mL | 4.0683 mL | |
| 10 mM | 0.2034 mL | 1.0171 mL | 2.0342 mL |