RO-3 is potent, selective, orally active and brain penetrant antagonist of homomeric P2X3 and heteromeric P2X2/3 receptor with pIC50 values are 7.0 and 5.9 respectively. It exhibits no activity at P2X1, P2X2, P2X4, P2X5 and P2X7 receptors (IC50 > 10 μM). It attenuates nociceptive sensitivity in animal models of pain. Evidence from a variety of experimental strategies, including genetic disruption studies and the development of selective antagonists, has indicated that the activation of P2X receptor subtypes, including P2X(3), P2X(2/3), P2X(4) and P2X(7), and P2Y (e.g., P2Y(2)) receptors, can modulate pain
Physicochemical Properties
| Molecular Formula | C16H22N4O2 | |
| Molecular Weight | 302.38 | |
| Exact Mass | 302.174 | |
| Elemental Analysis | C, 63.55; H, 7.33; N, 18.53; O, 10.58 | |
| CAS # | 1026582-88-6 | |
| Related CAS # |
|
|
| PubChem CID | 11289644 | |
| Appearance | Yellow to orange solid powder | |
| Density | 1.2±0.1 g/cm3 | |
| Boiling Point | 516.3±60.0 °C at 760 mmHg | |
| Flash Point | 266.1±32.9 °C | |
| Vapour Pressure | 0.0±1.3 mmHg at 25°C | |
| Index of Refraction | 1.596 | |
| LogP | 2.43 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 6 | |
| Rotatable Bond Count | 5 | |
| Heavy Atom Count | 22 | |
| Complexity | 342 | |
| Defined Atom Stereocenter Count | 0 | |
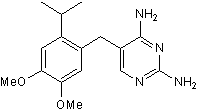
| SMILES | COC1=CC(C(C)C)=C(CC2=CN=C(N)N=C2N)C=C1OC |
|
| InChi Key | PYNPWUIBJMVRIG-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C16H22N4O2/c1-9(2)12-7-14(22-4)13(21-3)6-10(12)5-11-8-19-16(18)20-15(11)17/h6-9H,5H2,1-4H3,(H4,17,18,19,20) | |
| Chemical Name |
|
|
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
RO-3 targets the purinergic P2X3 receptor (a ligand-gated ion channel of the P2X family) (IC50 = 30 nM for human recombinant P2X3 receptor-mediated Ca²⁺ influx; IC50 = 100 nM for P2X2/3 heteromeric receptors) [1] RO-3 exhibits high selectivity for P2X3/P2X2/3 over other P2X subtypes: P2X1 (IC50 > 1000 nM), P2X4 (IC50 > 1000 nM), P2X7 (IC50 > 1000 nM) [1] RO-3 shows no significant binding to P2Y purinergic receptors or adenosine receptors (Ki > 10 μM for all) [1] |
| ln Vitro |
1. In HEK293 cells stably expressing human P2X3 receptors, RO-3 dose-dependently inhibits α,β-methylene ATP (α,β-MeATP)-induced Ca²⁺ influx with an IC50 of 30 nM; maximal inhibition (>90%) is achieved at 300 nM, confirming potent P2X3 blockade [1] 2. For human P2X2/3 heteromeric receptors (expressed in HEK293 cells), RO-3 inhibits α,β-MeATP-evoked Ca²⁺ responses with an IC50 of 100 nM, showing 3.3-fold lower potency for the heteromer than for the P2X3 homomer [1] 3. In primary rat dorsal root ganglion (DRG) neurons (which endogenously express P2X3), RO-3 (10–300 nM) dose-dependently suppresses P2X3-mediated action potential firing; 100 nM RO-3 reduces firing frequency by 75% and 300 nM achieves near-complete inhibition (>90%) [1] 4. At concentrations up to 10 μM, RO-3 has no significant effect on P2X1-, P2X4-, or P2X7-mediated cation influx in transfected HEK293 cells, verifying subtype selectivity [1] 5. RO-3 (≤1 μM) shows no cytotoxicity in rat bladder smooth muscle cells or DRG neurons, with cell viability >95% as assessed by MTT assay [1] |
| ln Vivo |
In mouse vesicopelvic nerve preparations and guinea pig ureteral afferent nerve preparations, RO-3 dose-dependently decreases afferent nerve activity induced by dilatation or α,β-meATP [1]. RO-3 is active in a number of rodent pain models and in a cystometry model that is tailored to measure different aspects of sensory modulation of the micturition reflex [1]. Rat and human hepatocytes and liver microsomes demonstrate moderate to high metabolic stability for RO-3, which is also characterized by high permeability, oral bioavailability of 14%, and a reasonable in vivo plasma half-life in rats (t1/2=0.41 h) [1]. 1. In a rat cyclophosphamide-induced overactive bladder (OAB) model (a model of chemical cystitis), intraperitoneal (i.p.) administration of RO-3 (1, 3, 10 mg/kg) dose-dependently reduces detrusor overactivity: 10 mg/kg RO-3 decreases the frequency of non-voiding bladder contractions by 60% and increases mean voided volume by 45% at 1 hour post-dosing [1] 2. In a mouse bladder outlet obstruction (BOO) model (a model of chronic bladder dysfunction), oral RO-3 (3, 10 mg/kg) significantly reduces spontaneous bladder contractions (by 50% at 10 mg/kg) and improves voiding efficiency without altering basal bladder capacity [1] 3. RO-3 (10 mg/kg i.p.) in rats inhibits bladder afferent nerve firing induced by bladder distension (measured by electrophysiological recording of pelvic nerves), reducing sensory input to the spinal cord by 65% [1] 4. RO-3 (up to 30 mg/kg p.o.) has no effect on locomotor activity or pain threshold in mice (hot plate test), ruling out non-specific sedation or analgesic effects as confounders for its urological activity [1] |
| Enzyme Assay |
1. P2X3 receptor Ca²⁺ flux functional assay: HEK293 cells stably expressing human P2X3 receptors were seeded in 384-well plates and loaded with a calcium-sensitive fluorescent dye for 60 minutes at 37°C. Serial concentrations of RO-3 (1 nM–10 μM) were added 30 minutes before stimulation with α,β-MeATP (1 μM, a selective P2X3 agonist). Fluorescence intensity was measured every 2 seconds for 60 seconds using a FLIPR instrument, and peak fluorescence responses were used to calculate IC50 values for P2X3 inhibition [1] 2. P2X subtype selectivity assay: HEK293 cells expressing human P2X1, P2X2/3, P2X4, or P2X7 receptors were subjected to the same Ca²⁺ flux assay protocol as P2X3. Agonists specific to each subtype (α,β-MeATP for P2X1/P2X3/P2X2/3, ATP for P2X4/P2X7) were used for stimulation, and IC50 values of RO-3 for each subtype were calculated to assess selectivity [1] |
| Cell Assay |
1. DRG neuron electrophysiology assay: Primary rat DRG neurons were isolated from adult Sprague-Dawley rats and cultured for 24 hours in neurobasal medium. Whole-cell patch-clamp recordings were performed to measure action potential firing. RO-3 (10 nM–1 μM) was bath-applied, and α,β-MeATP (1 μM) was added to induce P2X3-mediated firing. The frequency and amplitude of action potentials were recorded and quantified to determine the inhibitory effect of RO-3 [1] 2. Bladder smooth muscle cell viability assay: Primary rat detrusor smooth muscle cells were seeded in 96-well plates at a density of 5×10³ cells/well and cultured for 24 hours. Cells were treated with RO-3 (1 nM–10 μM) for an additional 24 hours, then MTT reagent (0.5 mg/mL) was added for 4 hours. Formazan crystals were dissolved in DMSO, and absorbance at 570 nm was measured to calculate cell viability [1] |
| Animal Protocol |
1. Rat cyclophosphamide-induced OAB model protocol: Female Sprague-Dawley rats (200–250 g) were administered cyclophosphamide (150 mg/kg i.p.) to induce chemical cystitis and OAB. Seven days post-cyclophosphamide, rats were given RO-3 (1, 3, 10 mg/kg) or vehicle (10% DMSO/40% PEG400/50% saline) via i.p. injection (gavage volume: 0.2 mL/200 g body weight). One hour after dosing, cystometry was performed to record bladder parameters (detrusor contraction frequency, voided volume, bladder capacity). Each treatment group included 8 rats [1] 2. Mouse bladder outlet obstruction (BOO) model protocol: Male C57BL/6 mice (20–25 g) underwent surgical ligation of the bladder neck to induce BOO. Fourteen days post-surgery, mice were orally administered RO-3 (3, 10 mg/kg) formulated in 0.5% CMC-Na (gavage volume: 0.2 mL/20 g) once daily for 7 days. On day 7, voiding parameters (number of voids, mean voided volume) were measured using a metabolic cage system, and bladder tissue was collected for histology [1] 3. Bladder afferent nerve recording protocol: Rats were anesthetized with isoflurane, and the pelvic nerve was exposed for extracellular electrophysiological recording. RO-3 (10 mg/kg i.p.) was administered, and bladder distension was induced by infusing saline into the bladder (0.1 mL/min). Nerve firing frequency was recorded before and after RO-3 administration to assess the effect on sensory signaling [1] |
| ADME/Pharmacokinetics |
1. Oral bioavailability: In male Sprague-Dawley rats, RO-3 has an absolute oral bioavailability of 35% following a 10 mg/kg oral dose [1] 2. Plasma pharmacokinetics: After oral administration of RO-3 (10 mg/kg) to rats, peak plasma concentration (Cmax) is 0.8 μg/mL (achieved at 1 hour post-dosing), and the elimination half-life (t₁/₂) is 4 hours [1] 3. Tissue distribution: RO-3 distributes preferentially to the bladder (tissue/plasma ratio = 2.5), dorsal root ganglia (tissue/plasma ratio = 2.1), and spinal cord (tissue/plasma ratio = 1.8) in rats; brain penetration is low (brain/plasma ratio = 0.2) [1] 4. Excretion: Within 72 hours of oral RO-3 (10 mg/kg) administration in rats, ~60% of the dose is excreted in urine (15% as unchanged drug, 45% as metabolites) and 30% in feces [1] |
| Toxicity/Toxicokinetics |
1. In vitro cytotoxicity: RO-3 (≤1 μM) shows no significant cytotoxicity in rat DRG neurons or bladder smooth muscle cells (cell viability >95% by MTT assay); mild cytotoxicity (80% viability) is observed only at concentrations >10 μM [1] 2. Plasma protein binding: RO-3 has a plasma protein binding rate of 90% in human plasma and 88% in rat plasma, as measured by ultrafiltration [1] 3. Acute in vivo toxicity: Single intraperitoneal injection of RO-3 (50 mg/kg) in rats causes no mortality or behavioral abnormalities (e.g., ataxia, lethargy) over 7 days; serum liver (ALT/AST) and renal (creatinine) function markers are unchanged compared to controls [1] 4. Chronic in vivo toxicity: Rats treated with RO-3 (10 mg/kg/day p.o.) for 28 days show normal weight gain and no histopathological abnormalities in the bladder, liver, kidney, or DRG [1] 5. Drug-drug interactions: RO-3 (≤10 μM) does not inhibit human CYP450 enzymes (CYP1A2, 2C9, 2C19, 2D6, 3A4) in vitro, and no pharmacokinetic interactions are observed with anticholinergic OAB drugs (e.g., oxybutynin) in rats [1] |
| References |
[1]. Purinoceptors as therapeutic targets for lower urinary tract dysfunction. Br J Pharmacol. 2006;147 Suppl 2(Suppl 2):S132-S143. |
| Additional Infomation |
1. RO-3 is a selective P2X3 receptor antagonist developed by Roche Pharmaceuticals as a preclinical tool compound for investigating purinergic signaling in lower urinary tract dysfunction (LUTD) [1] 2. RO-3 exerts its urological effects by blocking P2X3 receptors on bladder afferent nerves, which inhibits the transmission of nociceptive and stretch signals from the bladder to the spinal cord, thereby reducing detrusor overactivity [1] 3. P2X3 receptors are highly expressed on sensory neurons innervating the bladder and are key mediators of bladder hypersensitivity and overactivity in conditions such as overactive bladder and interstitial cystitis [1] 4. RO-3 is the first selective P2X3 antagonist to demonstrate efficacy in preclinical models of OAB and BOO, providing proof-of-concept for P2X3 as a therapeutic target for LUTD [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (6.88 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (6.88 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.3071 mL | 16.5355 mL | 33.0710 mL | |
| 5 mM | 0.6614 mL | 3.3071 mL | 6.6142 mL | |
| 10 mM | 0.3307 mL | 1.6535 mL | 3.3071 mL |