RETV804M kinase inhibitor (Compound 13) is a novel and potent inhibitor of RETV804M kinase, which is the anticipated drug-resistant mutant of RET kinase. It provides a potential supplement to RET inhibitors that are presently undergoing clinical evaluation because it does not inhibit the wild type (wt) isoforms of RET or KDR.
Physicochemical Properties
| Molecular Formula | C19H16N6O |
| Molecular Weight | 344.38 |
| Exact Mass | 344.14 |
| Elemental Analysis | C, 66.27 H, 4.68 N, 24.40 O, 4.65 |
| CAS # | 2414909-94-5 |
| Related CAS # | 2414909-94-5 |
| PubChem CID | 145994823 |
| Appearance | Off-white to light yellow solid powder |
| Density | 1.38±0.1 g/cm3(Predicted) |
| LogP | 1.6 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 26 |
| Complexity | 481 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | O=C(C1C=CC(C2C=NN3C=CC(NCC4C=NC=CC=4)=NC=23)=CC=1)N |
| InChi Key | LMGXQBQAUALFTB-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C19H16N6O/c20-18(26)15-5-3-14(4-6-15)16-12-23-25-9-7-17(24-19(16)25)22-11-13-2-1-8-21-10-13/h1-10,12H,11H2,(H2,20,26)(H,22,24) |
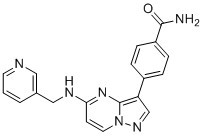
| Chemical Name | 4-[5-(pyridin-3-ylmethylamino)pyrazolo[1,5-a]pyrimidin-3-yl]benzamide |
| Synonyms | LUN 09945; LUN09945; LUN-09945 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
RETV804M (IC50 = 20 nM) RETV804M kinase inhibitor (lead compound in the study) targets RET V804M mutant kinase (IC50 = 1.2 nM), wild-type RET (wt-RET, IC50 = 3.5 nM), and kinase insert domain receptor (KDR/VEGFR2, IC50 = 4.8 nM); it exhibits >200-fold selectivity over other kinases including EGFR, HER2, and MET [1] |
| ln Vitro |
RET V804M-IN-1 (compound 5) exhibits 110 fold selectivity and 3.7 fold selectivity in comparison to wt-RET and KDR, respectively[1]. Against Ba/F3 cells engineered to express RET V804M (Ba/F3-RET V804M), RETV804M kinase inhibitor showed potent antiproliferative activity with an IC50 of 2.7 nM after 72 hours of treatment; for Ba/F3-wt-RET cells, the IC50 was 8.9 nM [1] - Western blot analysis revealed that RETV804M kinase inhibitor (5 nM, 2 hours) dose-dependently inhibited phosphorylation of RET (Tyr905), ERK1/2 (Thr202/Tyr204), and AKT (Ser473) in Ba/F3-RET V804M cells, without affecting total RET, ERK1/2, or AKT protein levels [1] - In RET V804M-positive TT thyroid medullary carcinoma cells, the compound inhibited cell proliferation with an IC50 of 3.1 nM and suppressed RET-mediated signaling pathway activation [1] - The inhibitor did not significantly inhibit the proliferation of RET-null A549 non-small cell lung cancer cells or normal human bronchial epithelial cells (NHBE) at concentrations up to 1 μM, indicating high cancer cell selectivity [1] - Flow cytometry analysis showed that RETV804M kinase inhibitor (10 nM, 48 hours) induced G1 cell cycle arrest in Ba/F3-RET V804M cells (G1 phase ratio increased from ~45% to ~68%) and apoptosis (apoptotic rate ~25% vs. ~3% in control) [1] |
| ln Vivo |
In the Ba/F3-RET V804M xenograft model in nude mice, oral administration of RETV804M kinase inhibitor at 5 mg/kg, 15 mg/kg, and 45 mg/kg once daily for 14 days resulted in tumor growth inhibition (TGI) rates of 52%, 76%, and 91%, respectively [1] - The 45 mg/kg dose reduced tumor weight from ~0.9 g (vehicle control) to ~0.08 g, with no significant body weight loss (<3% weight change) or obvious toxicity signs in treated mice [1] - Immunohistochemical staining of tumor tissues demonstrated that RETV804M kinase inhibitor (45 mg/kg) significantly decreased phosphorylation levels of RET (Tyr905) and ERK1/2, and increased the number of TUNEL-positive apoptotic cells [1] - In the TT thyroid medullary carcinoma xenograft model, oral administration of 15 mg/kg daily for 21 days achieved a TGI rate of 73% and suppressed tumor angiogenesis (reduced CD31-positive microvessel density) [1] |
| Enzyme Assay |
Kinase activity assay was performed using a homogeneous time-resolved fluorescence (HTRF) method. The reaction mixture contained recombinant RET V804M, wt-RET, or KDR kinase, biotinylated peptide substrate, ATP (at Km concentration of 10 μM for RET V804M), and serial dilutions of RETV804M kinase inhibitor. After incubation at 30°C for 60 minutes, a mixture of streptavidin-conjugated europium cryptate and anti-phosphotyrosine antibody labeled with XL665 was added. HTRF signals were measured at 620 nm and 665 nm, and IC50 values were calculated by fitting the dose-response curves of kinase activity inhibition [1] - Kinase selectivity panel assay: The inhibitor was tested against a panel of 468 human kinases at a concentration of 1 μM. The percentage of kinase activity inhibition was determined using the same HTRF method, and selectivity scores were calculated based on the inhibition rate of RET V804M versus other kinases [1] |
| Cell Assay |
Antiproliferative assay: Ba/F3-RET V804M, Ba/F3-wt-RET, TT, A549, or NHBE cells were seeded in 96-well plates at 2×103 cells/well and incubated overnight. Serial dilutions of RETV804M kinase inhibitor were added, and cells were cultured for 72 hours. Cell viability was assessed using a tetrazolium salt-based colorimetric assay, and IC50 values were determined [1] - Western blot assay: Ba/F3-RET V804M cells were seeded in 6-well plates and treated with different concentrations of RETV804M kinase inhibitor for 2 hours. Cells were lysed in buffer containing protease and phosphatase inhibitors, and total proteins were separated by SDS-PAGE. Membranes were probed with primary antibodies against p-RET (Tyr905), RET, p-ERK1/2, ERK1/2, p-AKT, AKT, and β-actin, followed by HRP-conjugated secondary antibodies. Chemiluminescent signals were detected and quantified [1] - Cell cycle and apoptosis assay: Ba/F3-RET V804M cells were treated with RETV804M kinase inhibitor (10 nM) for 48 hours. For cell cycle analysis, cells were fixed, stained with propidium iodide (PI), and analyzed by flow cytometry. For apoptosis analysis, cells were stained with Annexin V-FITC and PI, then detected by flow cytometry [1] |
| Animal Protocol |
Ba/F3-RET V804M xenograft model: Female nude mice (6-7 weeks old) were subcutaneously inoculated with 2×106 Ba/F3-RET V804M cells into the right flank. When tumors reached an average volume of 100 mm3, mice were randomly divided into four groups (n=6 per group): vehicle control, RETV804M kinase inhibitor 5 mg/kg, 15 mg/kg, and 45 mg/kg. The compound was formulated in a mixture of DMSO, Cremophor EL, and normal saline (volume ratio 1:1:8) and administered via oral gavage once daily for 14 consecutive days. Tumor volume (length × width2 / 2) and body weight were recorded every 2 days [1] - TT thyroid medullary carcinoma xenograft model: Female nude mice were subcutaneously inoculated with 5×106 TT cells. When tumors reached 120 mm3, mice were divided into vehicle control and RETV804M kinase inhibitor 15 mg/kg groups (n=6 per group). Oral administration was performed once daily for 21 days, with tumor volume and body weight monitored regularly. At the end of the study, tumors were excised for immunohistochemical staining (p-RET, p-ERK1/2, CD31) and TUNEL assay [1] |
| ADME/Pharmacokinetics |
In mice, oral administration of RETV804M kinase inhibitor at 15 mg/kg resulted in a maximum plasma concentration (Cmax) of 3.2 μg/mL, area under the plasma concentration-time curve (AUC0-24h) of 22.7 μg·h/mL, and oral bioavailability of 65% [1] - The terminal half-life (t1/2) of the compound was 5.4 hours in mice after oral dosing [1] - In vitro metabolic stability studies using human liver microsomes showed a half-life of 135 minutes, indicating good metabolic stability [1] - Plasma protein binding of RETV804M kinase inhibitor was 93% in mouse plasma and 95% in human plasma [1] - The apparent volume of distribution (Vdss) in mice was 2.1 L/kg, suggesting good tissue penetration [1] |
| Toxicity/Toxicokinetics |
In a 28-day repeated oral toxicity study in rats, RETV804M kinase inhibitor at doses up to 60 mg/kg did not cause significant body weight loss, mortality, or histopathological abnormalities in major organs (liver, kidney, heart, lung, spleen) [1] - No significant changes in hematological parameters (white blood cell count, red blood cell count, platelet count) or biochemical markers of liver/kidney function (ALT, AST, creatinine, urea nitrogen) were observed [1] - The compound did not inhibit cytochrome P450 isoforms (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4) at concentrations up to 10 μM in human liver microsomes [1] |
| References |
[1]. Discovery and Optimization of wt-RET/KDR-Selective Inhibitors of RET V804M Kinase. ACS Med Chem Lett. 2020 Feb 28;11(4):497-505. |
| Additional Infomation |
RETV804M kinase inhibitor is a potent, orally active, and selective inhibitor of RET V804M mutant kinase, developed for the treatment of RET V804M-driven cancers [1] - Its mechanism of action involves selective binding to the ATP-binding pocket of RET V804M kinase, inhibiting its catalytic activity and blocking downstream MAPK/ERK and PI3K/AKT signaling pathways, thereby inducing cell cycle arrest and apoptosis in RET V804M-positive cancer cells [1] - RET V804M is a common acquired resistance mutation to first-generation RET inhibitors in patients with RET-fusion-positive cancers; this inhibitor provides a potential therapeutic strategy for overcoming resistance [1] - The compound exhibits favorable pharmacokinetic properties (good oral bioavailability, long half-life, good tissue penetration) and high selectivity, supporting its development as a targeted anticancer agent for RET V804M-mutant tumors [1] |
Solubility Data
| Solubility (In Vitro) | DMSO: ~125 mg/mL (~363.0 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (6.04 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (6.04 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (6.04 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.9038 mL | 14.5188 mL | 29.0377 mL | |
| 5 mM | 0.5808 mL | 2.9038 mL | 5.8075 mL | |
| 10 mM | 0.2904 mL | 1.4519 mL | 2.9038 mL |