RAF709 is a novel and potent inhibitor of the Raf kinase B/C isoforms (compound example 131 from patent WO2014151616A1) developed through a hypothesis-driven approach focusing on drug-like properties. For c-Raf and b-Raf, the IC50 values are, respectively, 0.5 and 1.8 nM. It is effective in a KRAS mutant xenograft model, soluble, and kinase selective, and it demonstrated a high level of selectivity over other kinases. Additionally, RAF709 showed at 1 μM that it bound to BRAF, BRAFV600E, and CRAF with greater than 99% on-target affinity and little to no off-target affinity for DDR1 (>99%), DDR2 (86%), FRK (92%), and PDGFRb (96%). When RAF709 and a MEK inhibitor are used together, KRASmut tumors exhibit improved antitumor activity.
Physicochemical Properties
| Molecular Formula | C28H29F3N4O4 | |
| Molecular Weight | 542.55 | |
| Exact Mass | 542.214 | |
| Elemental Analysis | C, 61.99; H, 5.39; F, 10.50; N, 10.33; O, 11.80 | |
| CAS # | 1628838-42-5 | |
| Related CAS # |
|
|
| PubChem CID | 90408826 | |
| Appearance | White to off-white solid powder | |
| LogP | 4 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 10 | |
| Rotatable Bond Count | 6 | |
| Heavy Atom Count | 39 | |
| Complexity | 793 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | FC(C1=C([H])C([H])=C([H])C(=C1[H])C(N([H])C1C([H])=NC(C([H])([H])[H])=C(C=1[H])C1C([H])=NC(=C(C=1[H])N1C([H])([H])C([H])([H])OC([H])([H])C1([H])[H])OC1([H])C([H])([H])C([H])([H])OC([H])([H])C1([H])[H])=O)(F)F |
|
| InChi Key | FYNMINFUAIDIFL-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C28H29F3N4O4/c1-18-24(15-22(17-32-18)34-26(36)19-3-2-4-21(13-19)28(29,30)31)20-14-25(35-7-11-38-12-8-35)27(33-16-20)39-23-5-9-37-10-6-23/h2-4,13-17,23H,5-12H2,1H3,(H,34,36) | |
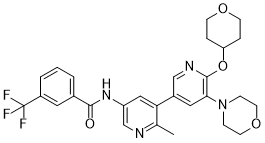
| Chemical Name | N-[6-methyl-5-[5-morpholin-4-yl-6-(oxan-4-yloxy)pyridin-3-yl]pyridin-3-yl]-3-(trifluoromethyl)benzamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
C-Raf (IC50 = 0.4 nM); BRAF(V600E) (IC50 = 1 nM); B-Raf (IC50 = 1.5 nM)
RAF709 is a potent and selective inhibitor of RAF family kinases, including BRAF (wild-type and V600E mutant), CRAF, and ARAF; IC50 values are as follows: BRAF V600E (1.2 nM), wild-type BRAF (3.5 nM), CRAF (2.8 nM), ARAF (5.1 nM) [determined by recombinant kinase activity assay]. [1] |
| ln Vitro |
RAF709 stabilizes BRAF-CRAF dimers with an EC50 of 0.8 μM. The cellular assays used to measure the dose-response of pMEK and pERK in Calu-6 cells used EC50 values of 0.02 and 0.1 μM with minimal paradoxical activation and 0.95 M with proliferative inhibition[1]. 1. RAF kinase activity inhibition: RAF709 potently inhibited the kinase activity of recombinant BRAF V600E, wild-type BRAF, CRAF, and ARAF in a dose-dependent manner, with IC50 values in the low nanomolar range (1.2–5.1 nM). It showed >1000-fold selectivity over 450+ other kinases (e.g., EGFR, ALK, MET, PI3K) at 1 μM, confirming high kinome selectivity. [1] 2. Antiproliferative activity against RAS pathway-mutant cancer cells: RAF709 exhibited potent antiproliferative effects on cancer cell lines harboring BRAF V600E (A375, SK-MEL-28, Colo205), KRAS (HCT116, SW620, Panc-1), or NRAS (WM1346, SK-MEL-1) mutations, with GI50 values ranging from 4.3 nM to 28.7 nM. No significant antiproliferative activity was observed in RAS/RAF wild-type cell lines (e.g., MCF-7, HEK293) at concentrations up to 1 μM. [1] 3. Inhibition of MEK/ERK signaling pathway: RAF709 (10–100 nM) dose-dependently suppressed the phosphorylation of MEK1/2 and ERK1/2 in A375 (BRAF V600E) and HCT116 (KRAS G13D) cells (Western blot analysis). The inhibition was sustained for 24 hours after a single treatment, with p-ERK levels reduced by >80% at 50 nM. [1] 4. Induction of apoptosis in RAS-mutant cancer cells: RAF709 (50 nM) induced apoptosis in A375 and SW620 cells, as evidenced by increased Annexin V-FITC/PI double-positive cells (28.3% and 21.7% vs. 4.2% and 3.8% in vehicle controls) and upregulation of Cleaved-Caspase 3/7 and PARP cleavage (Western blot). [1] 5. Suppression of colony formation: RAF709 (10–50 nM) dose-dependently inhibited colony formation of A375 and HCT116 cells, with colony numbers reduced by 52% and 47% at 30 nM, respectively, compared to vehicle controls. [1] |
| ln Vivo |
RAF709 proves to be soluble, kinase selective, and efficacious in a KRAS mutant xenograft model. In Calu-6 tumors, RAF709 causes a dose-dependent inhibition of pERK and dose-proportional increases in plasma exposure. Treatment with RAF709 produces dose-dependent antitumor activity, with 10 mg/kg being subefficacious (%T/C=92%), 30 mg/kg producing measurable antitumor activity (%T/C=46%), and 200 mg/kg producing mean tumor regression of 92%, whereas the same high dose is ineffective in the PC3, KRAS WT model[1]. 1. Antitumor activity in BRAF V600E-mutant A375 melanoma xenograft model: [1] - Female nu/nu mice bearing subcutaneous A375 tumors (100–150 mm³) were orally administered RAF709 at 10, 30, or 100 mg/kg once daily (QD) for 21 days. - RAF709 exhibited dose-dependent tumor growth inhibition (TGI): 10 mg/kg (TGI=58%), 30 mg/kg (TGI=83%), 100 mg/kg (TGI=92%). Tumor regression was observed in 4/6 mice at 100 mg/kg. - Pharmacodynamic (PD) analysis of tumor tissues showed that RAF709 (30 mg/kg) reduced p-ERK expression by 78% at 6 hours post-administration, with sustained inhibition (>60%) for 24 hours. 2. Antitumor activity in KRAS-mutant HCT116 colorectal cancer xenograft model: [1] - Female nu/nu mice bearing subcutaneous HCT116 tumors were orally administered RAF709 at 30 or 60 mg/kg QD for 21 days. - TGI rates were 71% (30 mg/kg) and 85% (60 mg/kg), with no significant body weight loss (<5%) in either group. - Tumor tissue immunohistochemistry (IHC) confirmed reduced Ki-67 (proliferation marker) expression and increased TUNEL-positive (apoptotic) cells. 3. Antitumor activity in NRAS-mutant WM1346 melanoma xenograft model: [1] - Oral administration of RAF709 (30 mg/kg QD for 21 days) resulted in TGI of 76%, with significant reduction in p-MEK and p-ERK levels in tumor tissues. |
| Enzyme Assay |
The MEK1 protein substrate, which carries the K97R mutation, was used as the substrate for the CRAF kinase assay along with 10 nM kinase-dead MEK1, 3 μM ATP, and 10 pM CRAF Y340E/Y341E. The reaction buffer contained 1 mM DTT, 50 mM Tris pH 7.5, 10 μL MgCl2, 0.05% BSA, 50 mM NaCl, and 0.01% Tween-20. The reactions took place in white 384 shallow well plates with a volume of 10 μL at room temperature for 40 minutes before being quenched with a solution of 5 μL per well (50 mM Tris pH 7.5, 50 mM EDTA). Reactions that had reached their end were given 5 μL/well of detection reagents, which included 50 mM Tris pH 7.5, 0.01% Tween-20, antiphospho MEK1/2 S217/S221 antibody diluted 1:1,000, 0.01 mg/mL of AlphaScreen Protein A-coated acceptor beads, and 0.01 mg/mL of streptavidin-coated donor beads. After a night of incubation at room temperature, plates were read in an EnVision plate reader. Compounds were tested in 16-point, 3-fold format in compound inhibition studies over a concentration range of 25 μM to 1.74 × 10−6 μM. The final concentration of DMSO was 0.5%. Before adding substrates to begin the reaction, compounds were given a 30-minute preincubation with CRAF. The IC50 of the compounds was determined by fitting the data on inhibition to a four-parameter logistic equation. 1. Recombinant RAF kinase activity assay (HTRF-based): [1] Recombinant human RAF kinases (BRAF V600E, wild-type BRAF, CRAF, ARAF) were diluted in assay buffer containing MgCl₂ and DTT. The reaction mixture included kinase substrate (a biotinylated MEK1-derived peptide), ATP (at Km concentration), and serial concentrations of RAF709. After incubation at 37°C for 60 minutes, the reaction was stopped by adding EDTA-containing buffer. Streptavidin-conjugated Europium cryptate and anti-phospho-MEK antibody labeled with XL665 were added to detect phosphorylated substrate via HTRF. The fluorescence resonance energy transfer signal was measured, and IC50 values were calculated by fitting the dose-response curves using nonlinear regression. 2. Kinome selectivity assay: [1] RAF709 was tested at 1 μM against a panel of 468 human kinases using a radiometric filter-binding assay. Kinase activity was measured by the incorporation of radioactive ATP into specific substrates. The inhibition rate of each kinase was calculated relative to vehicle control, and selectivity was defined as the ratio of IC50 for non-RAF kinases to IC50 for BRAF V600E. [1] |
| Cell Assay |
For 1 hour, HCT116 cells are treated with DMSO or RAF709 at the indicated concentrations; a comparison treatment with 1 mmol/L dabrafenib is also included. BRAF or CRAF is immunoprecipitated, and the BRAF and CRAF proteins are then analyzed on a Western blot to determine whether BRAF/CRAF dimerization has occurred. Western blot analysis is used to quantify the amounts of pMEK and pERK in whole-cell lysates (WCL). GAPDH level is used as a loading control. 1. Cell proliferation (GI50) assay: [1] RAS/RAF-mutant and wild-type cancer cell lines were seeded in 96-well plates at 3×10³–5×10³ cells/well and incubated overnight. Serial concentrations of RAF709 (0.1 nM–1 μM) were added, and cells were cultured for 72 hours. Cell viability was assessed by adding a tetrazolium-based reagent, and absorbance was measured at 570 nm. GI50 values (concentration inhibiting cell growth by 50%) were calculated using graphing software. 2. Western blot analysis for MEK/ERK pathway inhibition: [1] Cancer cells (A375, HCT116) were seeded in 6-well plates and treated with RAF709 (10–100 nM) for 24 hours. Cells were lysed in RIPA buffer with protease and phosphatase inhibitors, and protein concentrations were determined by BCA assay. Equal amounts of protein were separated by SDS-PAGE, transferred to PVDF membranes, and probed with primary antibodies against p-RAF, p-MEK1/2, MEK1/2, p-ERK1/2, ERK1/2, Cleaved-Caspase 3, Cleaved-PARP, and β-actin. HRP-conjugated secondary antibodies and ECL substrate were used for protein detection. 3. Apoptosis assay (Annexin V-FITC/PI staining): [1] A375 and SW620 cells were treated with RAF709 (50 nM) for 48 hours. Cells were harvested, washed with PBS, and resuspended in binding buffer. Annexin V-FITC and PI were added, and cells were incubated in the dark for 15 minutes. Apoptotic cells (Annexin V-positive/PI-negative and Annexin V-positive/PI-positive) were quantified by flow cytometry. 4. Colony formation assay: [1] Cancer cells were seeded in 6-well plates at 500 cells/well and incubated overnight. RAF709 (10–50 nM) was added, and cells were cultured for 10–14 days. Colonies were fixed with formalin, stained with crystal violet, and counted under a microscope. The colony formation rate was calculated relative to vehicle control. [1] |
| Animal Protocol |
Calu-6 model (tumor bearing mice) 10, 30, or 200 mg/kg oral administration 1. Xenograft tumor models (A375, HCT116, WM1346): [1] - Animals: Female nu/nu nude mice (6–8 weeks old) were housed under SPF conditions. - Tumor inoculation: 5×10⁶ (A375, WM1346) or 1×10⁷ (HCT116) cells suspended in Matrigel:PBS (1:1) were subcutaneously injected into the right flank of each mouse. - Grouping and drug administration: When tumors reached 100–150 mm³, mice were randomly divided into vehicle control and RAF709 treatment groups (n=6 per group). RAF709 was dissolved in 0.5% methylcellulose + 0.2% Tween 80 and administered via oral gavage at doses of 10, 30, 60, or 100 mg/kg once daily for 21 days. Vehicle control received the same volume of solvent. - Tumor and body weight monitoring: Tumor volume (V = length×width²/2) and body weight were measured every 3 days. - Sample collection: At the end of treatment, mice were euthanized, and tumors were excised, weighed, and snap-frozen in liquid nitrogen for Western blot analysis or fixed in formalin for IHC staining. - PD analysis: Tumor tissues were lysed for Western blot detection of p-MEK, p-ERK, and apoptotic markers; formalin-fixed tissues were paraffin-embedded, sectioned, and stained with Ki-67 antibody or TUNEL kit. [1] |
| ADME/Pharmacokinetics |
1. Oral bioavailability: In CD-1 mice, oral administration of RAF709 (30 mg/kg) resulted in an oral bioavailability (F) of 82%, with Cmax = 1.2 μg/mL and AUC₀–24h = 6.8 μg·h/mL. [1] 2. Half-life: Terminal half-life (t1/2) in mice was 5.7 hours (oral) and 4.3 hours (intravenous, 10 mg/kg). [1] 3. Tissue distribution: After oral administration (30 mg/kg), RAF709 distributed widely to tissues, with tumor-to-plasma concentration ratio of 2.3 at 6 hours post-dosing. [1] 4. Metabolism: In vitro liver microsomal stability assays showed that RAF709 was primarily metabolized by CYP3A4, with minimal metabolism by other CYP isoforms (CYP1A2, 2C9, 2C19, 2D6). [1] 5. Excretion: In mice, 72-hour excretion data showed 68% of the dose was excreted in feces (52% as parent drug) and 12% in urine (3% as parent drug). [1] 6. Plasma protein binding: In human plasma, RAF709 had a plasma protein binding rate of 95% (determined by equilibrium dialysis). [1] |
| Toxicity/Toxicokinetics |
1. Acute toxicity: Single oral administration of RAF709 up to 200 mg/kg in CD-1 mice caused no mortality or significant toxicity (body weight loss <10%, no abnormal clinical signs). [1] 2. Subchronic toxicity: Repeat oral administration (30 mg/kg QD for 28 days) in mice showed no significant changes in hematological parameters (WBC, RBC, platelets), clinical chemistry (ALT, AST, BUN, creatinine), or organ weights (liver, kidney, heart, spleen). Histopathological examination of major organs revealed no drug-related lesions. [1] 3. No drug-drug interaction potential: In vitro CYP inhibition assays showed RAF709 did not inhibit CYP1A2, 2C9, 2C19, 2D6, or 3A4 at concentrations up to 10 μM. [1] |
| References |
[1]. Design and Discovery of N-(2-Methyl-5'-morpholino-6'-((tetrahydro-2H-pyran-4-yl)oxy)-[3,3'-bipyridin]-5-yl)-3-(trifluoromethyl)benzamide (RAF709): A Potent, Selective, and Efficacious RAF Inhibitor Targeting RAS Mutant Cancers. J Med. |
| Additional Infomation |
1. Chemical structure: RAF709 (chemical name: N-(2-Methyl-5'-morpholino-6'-((tetrahydro-2H-pyran-4-yl)oxy)-[3,3'-bipyridin]-5-yl)-3-(trifluoromethyl)benzamide) is a small-molecule inhibitor belonging to the bipyridine amide class. [1] 2. Background: Aberrant activation of the RAS-RAF-MEK-ERK signaling pathway is a key driver of multiple cancers, including melanoma, colorectal cancer, and pancreatic cancer. Mutations in BRAF (V600E), KRAS, or NRAS lead to constitutive pathway activation, promoting tumor proliferation, survival, and metastasis. [1] 3. Mechanism of action: RAF709 competitively binds to the ATP-binding pocket of RAF family kinases (BRAF, CRAF, ARAF), inhibiting their kinase activity and blocking downstream MEK/ERK signaling. This leads to suppression of tumor cell proliferation, induction of apoptosis, and inhibition of tumor growth. [1] 4. Selectivity: RAF709 exhibits high selectivity for RAF kinases over other kinases (selectivity score >0.95 at 1 μM), minimizing off-target effects. [1] 5. Therapeutic potential: Preclinical data demonstrate that RAF709 is efficacious against a broad range of RAS pathway-mutant cancers (BRAF V600E, KRAS, NRAS), supporting its potential as a targeted therapy for patients with RAS/RAF-mutant solid tumors. [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (4.61 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (4.61 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (4.61 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8431 mL | 9.2157 mL | 18.4315 mL | |
| 5 mM | 0.3686 mL | 1.8431 mL | 3.6863 mL | |
| 10 mM | 0.1843 mL | 0.9216 mL | 1.8431 mL |