Physicochemical Properties
| Molecular Formula | C26H26N6O2 |
| Molecular Weight | 454.523644924164 |
| Exact Mass | 454.211 |
| CAS # | 1800505-64-9 |
| PubChem CID | 102004343 |
| Appearance | White to yellow solid powder |
| LogP | 4.3 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 34 |
| Complexity | 706 |
| Defined Atom Stereocenter Count | 0 |
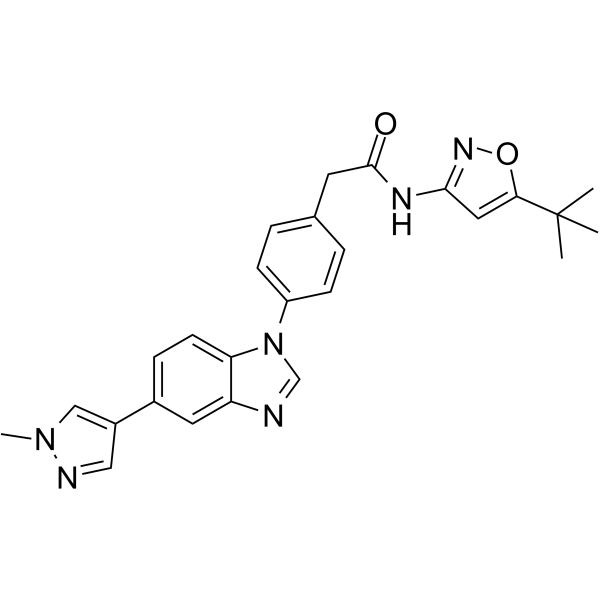
| SMILES | O1C(=CC(=N1)NC(CC1C=CC(=CC=1)N1C=NC2C=C(C3C=NN(C)C=3)C=CC1=2)=O)C(C)(C)C |
| InChi Key | NJLMIILZNLZZFW-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C26H26N6O2/c1-26(2,3)23-13-24(30-34-23)29-25(33)11-17-5-8-20(9-6-17)32-16-27-21-12-18(7-10-22(21)32)19-14-28-31(4)15-19/h5-10,12-16H,11H2,1-4H3,(H,29,30,33) |
| Chemical Name | N-(5-tert-butyl-1,2-oxazol-3-yl)-2-[4-[5-(1-methylpyrazol-4-yl)benzimidazol-1-yl]phenyl]acetamide |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Pz-1 is a type II tyrosine kinase inhibitor that attaches itself to the kinase's DFG-out conformation. Tyrosine phosphorylation of VEGFR2 and clinically relevant RET mutants, such as vandetanib- and cabozantinib-resistant mutants (RETV804M and RETV804L), was significantly inhibited by 1.0 nM Pz-1 in a cell-based assay [1]. |
| ln Vitro |
Pz-1 is a type II tyrosine kinase inhibitor that attaches itself to the kinase's DFG-out conformation. Tyrosine phosphorylation of VEGFR2 and clinically relevant RET mutants, such as vandetanib- and cabozantinib-resistant mutants (RETV804M and RETV804L), was significantly inhibited by 1.0 nM Pz-1 in a cell-based assay [1]. In a biochemical kinase assay, Pz-1 exhibited potent inhibitory activity against wild-type RET, VEGFR2, and clinically relevant RET mutants (RETC634R, RETM918T, RETV804M, RETV804L) with IC50 values all less than 1.0 nM. [1] In cell-based assays using transfected HEK293 cells, a 1.0 nM dose of Pz-1 strongly inhibited the tyrosine phosphorylation of RET oncoproteins (RETC634R, RETM918T, RETV804M, RETV804L). [1] In VEGFA-stimulated HEK293 cells transfected with VEGFR2, 1.0 nM Pz-1 effectively inhibited VEGFR2 phosphorylation. Similarly, in human umbilical vein endothelial cells (HUVECs), Pz-1 inhibited endogenous VEGFA-induced VEGFR2 phosphorylation. [1] In NIH3T3 fibroblasts transformed with RETC634Y, Pz-1 inhibited cell proliferation with an IC50 of 0.5 nM. In contrast, the IC50 for inhibiting proliferation of NIH3T3 cells transformed with HRasG12V was 34.4 nM. Pz-1 did not exert detectable growth inhibition in parental NIH3T3 cells at concentrations up to 100.0 nM. [1] A kinome-wide selectivity screen against a panel of 91 kinases at 50.0 nM concentration showed that Pz-1 was active on only 7 additional kinases besides RET and VEGFR2, indicating good global selectivity. [1] |
| ln Vivo |
Pz-1 binds actively to VEGFR2, preventing the blood flow that RET needs to promote growth. When taken orally at 1.0 mg/kg per day, Pz-1 prevented the phosphorylation of RET and VEGFR2 in tumor tissues and eliminated the tumor development caused by RET mutant fibroblasts. Up to 100.0 mg/kg, Pz-1 exhibits no discernible toxicity, indicating a broad therapeutic window [1]. In immunodeficient (nu/nu) mice injected with NIH3T3 RETC634Y cells, daily oral administration of Pz-1 at 1.0 mg/kg/day, initiated before tumor appearance, completely prevented the formation of RET-driven tumors. [1] In mice injected with NIH3T3 HRasG12V cells, the same treatment (1.0, 3.0, or 10.0 mg/kg/day) reduced but did not abrogate tumor formation, demonstrating preferential efficacy for RET-driven tumors. [1] Analysis of tumor tissues showed that Pz-1 (1.0 mg/kg) inhibited the phosphorylation of both RET and VEGFR2 in RET-driven tumors. It also inhibited downstream MAPK and mTOR signaling cascades specifically in RET-driven tumors, but not in Ras-driven tumors. However, VEGFR2 phosphorylation was inhibited in tumors driven by both RET and Ras. [1] |
| Enzyme Assay |
A microfluidics separation-based biochemical kinase assay was used to determine IC50 values. The assay conditions included a specific ATP concentration (e.g., 190 µM as noted for some compounds). Inhibitory activity was measured by assessing the reduction in kinase activity towards its substrate. The data presented are means from at least three independent experiments. [1] For the determination of the ATP Km for RET, standard enzymatic kinetics methods were employed, resulting in a Km value of 12.00 ± 0.26 µM. To characterize the type of inhibition, the IC50 of a precursor compound (3b) was determined at varying ATP concentrations (6.2, 12.5, 50.0, and 100.0 µM), showing that inhibition was partially competitive with ATP. [1] Dissociation constant (Kd) values for Pz-1 and key precursors against RET, VEGFR2, and the RETV804M mutant were determined using an active site-directed competition binding assay. [1] |
| Cell Assay |
To assess inhibition of oncogenic RET phosphorylation, cells expressing specific RET mutants were treated with Pz-1 or vehicle. After treatment, cells were lysed, and proteins were resolved by SDS-PAGE. Phosphorylation levels of RET at specific tyrosine residues (e.g., Y1062) were analyzed by western blotting using phospho-specific antibodies, with total RET protein levels serving as a loading control. [1] To evaluate inhibition of VEGFR2 phosphorylation, HEK293 cells transfected with VEGFR2 or native HUVECs were serum-starved, stimulated with VEGFA in the presence or absence of Pz-1, and then lysed. Phosphorylation of VEGFR2 at Y1175 was detected by western blotting. [1] Anti-proliferative activity was measured using cell viability assays. NIH3T3 cells transformed with RETC634Y or HRasG12V, as well as parental NIH3T3 cells, were treated with a range of Pz-1 concentrations. After an appropriate incubation period, cell viability or proliferation was quantified, and IC50 values were calculated. [1] |
| Animal Protocol |
For the in vivo efficacy study, immunodeficient (nu/nu) mice were subcutaneously injected with NIH3T3 fibroblasts transformed by either RETC634Y or HRasG12V. Treatment was initiated before palpable tumors formed. Pz-1 was administered daily by oral gavage (per os, PO) at doses of 1.0, 3.0, or 10.0 mg/kg/day. Control groups received vehicle only. Tumor growth was monitored over time by measuring tumor volume. [1] For toxicity assessment, mice were treated with Pz-1 at daily doses up to 100.0 mg/kg for one week, and body weight and serum alanine transaminase (ALT) levels were monitored. [1] |
| ADME/Pharmacokinetics | The literature states that Pz-1 displayed highly favorable pharmacokinetic properties, but no specific parameters (e.g., half-life, bioavailability, AUC) are provided in the main text or accessible supplementary information. [1] |
| Toxicity/Toxicokinetics | In a one-week study in mice, Pz-1 was highly tolerated at daily oral doses up to 100.0 mg/kg with no detectable signs of toxicity. Serum alanine transaminase (ALT) levels increased in a linear fashion with dose but remained within the normal range, serving as a monitorable marker. No other specific toxicities (e.g., hematological, renal) or lethal doses are reported. [1] |
| References |
[1]. Fragment-Based Discovery of a Dual pan-RET/VEGFR2 Kinase Inhibitor Optimized for Single-Agent Polypharmacology. Angew Chem Int Ed Engl. 2015 Jul 20;54(30):8717-21. |
| Additional Infomation |
Pz-1 was discovered via a fragment-based screen using a kinase-directed fragment (KDF) library, followed by optimization guided by computational modeling and structure-activity relationship (SAR) studies. [1] It is characterized as a Type-II tyrosine kinase inhibitor that binds to the DFG-out conformation of the kinase. Molecular modeling suggests its equipotency on RET and VEGFR2 is facilitated by free rotation of a methylene linker, allowing different binding geometries in the two kinases. [1] Pz-1 retains potent activity against RET gatekeeper mutants (V804M/L), which are refractory to the approved drugs vandetanib and cabozantinib. [1] The study proposes a single-agent polypharmacology strategy, where Pz-1 simultaneously targets the tumor parenchyma (via RET inhibition) and the tumor stroma (via anti-angiogenic VEGFR2 inhibition). [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~50 mg/mL (~110.01 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.50 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.50 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.50 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2001 mL | 11.0006 mL | 22.0012 mL | |
| 5 mM | 0.4400 mL | 2.2001 mL | 4.4002 mL | |
| 10 mM | 0.2200 mL | 1.1001 mL | 2.2001 mL |