Physicochemical Properties
| Molecular Formula | C13H18NNAO4S |
| Molecular Weight | 307.34 |
| Exact Mass | 307.08542 |
| CAS # | 23795-03-1 |
| Related CAS # | Probenecid-d14;1189657-87-1;Probenecid;57-66-9;Probenecid-d7;2012598-90-0 |
| PubChem CID | 23662399 |
| Appearance | White to off-white solids at room temperature |
| Boiling Point | 438ºC at 760mmHg |
| Flash Point | 218.7ºC |
| Vapour Pressure | 1.91E-08mmHg at 25°C |
| LogP | 1.942 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 20 |
| Complexity | 380 |
| Defined Atom Stereocenter Count | 0 |
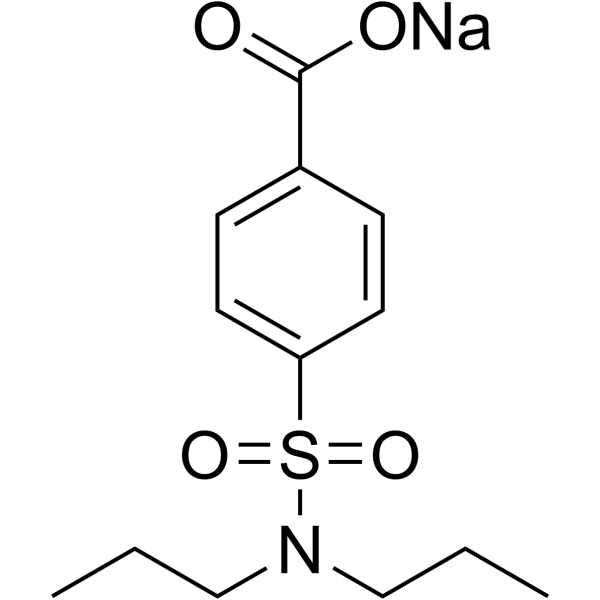
| SMILES | [Na+].CCCN(S(C1C=CC(C([O-])=O)=CC=1)(=O)=O)CCC |
| InChi Key | QCCCFHDTBTUDEA-UHFFFAOYSA-M |
| InChi Code | InChI=1S/C13H19NO4S.Na/c1-3-9-14(10-4-2)19(17,18)12-7-5-11(6-8-12)13(15)16;/h5-8H,3-4,9-10H2,1-2H3,(H,15,16);/q;+1/p-1 |
| Chemical Name | sodium 4-(dipropylsulfamoyl)benzoate |
| Synonyms | Probenesulfonate; Probenecid sodium; 23795-03-1; Sodium probenecid; Benemid, sodium salt; Probenecid sodium salt; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | OAT1/OAT3; Pannexin-1 (Panx1) ; MRP1; transient receptor potential vanilloid 2 (TRPV2) |
| ln Vitro | Probenecid potently inhibits the absorption of ATP-dependent active vesicles N-ethylmaleimide glutathione (NEM-GS) by MRP1 and MRP2. Significant inhibition of MRP1-ATPase was seen at increasing organic anion concentrations. The ATPase activity of MRP2 is influenced by probenecid (about KACT=250 μM), sulfinpyrazone (KACT=300 μM) and indomethacin (KACT=150 μM), and the ATPase activation is even stronger than that of NEM-GS. Activation of MRP2-ATPase by organic anions follows a bell-shaped curve, with maximum values at 2 mM for Probenecid, 800 μM for sulfinpyrazone, and 400 μM for indomethacin [2]. Probenecid is an inhibitor of hTAS2R16, hTAS2R38 and hTAS2R43 bitter taste receptors. Probenecid operates on a subset of TAS2R and suppresses through a new allosteric mechanism of action. Probenecid is also often utilized to improve cell signaling in GPCR calcium mobilization experiments. Probenecid specifically suppresses cellular responses mediated by the bitter taste receptor hTAS2R16 and provides molecular and pharmacological evidence for direct contact with this GPCR using a non-competitive (allosteric) mechanism [3]. |
| ln Vivo | When compared to control mice fed saline, probenecid increased the contractility of WT mice as indicated by their ejection fraction (EF). At all doses of 75 mg/kg and higher, increased contractility was observed within 5 minutes of the bolus (peak changes at 75 mg/kg, 100 mg/kg, and 200 mg/kg were 5.26±3.35, 8.40±2.80, and 7.32± 2.52, respectively). With an estimated EC50 of 49.33 mg/kg, changes in contractility assessed at 5-minute intervals for a total of 30 minutes showed a dose-dependent increase in contractility. EF stayed raised for at least an hour in patients examined over a longer duration (n=5 at 200 mg/kg IV) (mean increase in EF from baseline 8.9±2.57) [1]. |
| Animal Protocol | In order to obtain a dose response curve, male C57 WT (n=39) mice 12-16 weeks of age are anesthetized with isoflurane while intravenous jugular access (IV) is obtained under a microscope. Subsequently, an echocardiographic study with both M-mode and B-mode is obtained in parasternal long axis (PSLAX) as described below. Either saline or different doses of Probenecid (increasing from 2 to 200mg/kg) are injected (bolus IV) for the initial contractility studies in WT mice. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Excreted principally in the urine as monoacyl glucuronide and unchanged drug. Alkalinization of urine increases renal probenecid excretion. PROBENECID IS COMPLETELY ABSORBED AFTER ORAL ADMIN. PEAK PLASMA CONCN ARE REACHED IN 2-4 HR. THE HALF-LIFE OF THE DRUG IN PLASMA IS DOSE DEPENDENT AND VARIES FROM LESS THAN 5 HR TO MORE THAN 8 HR. BETWEEN 85 & 95% OF DRUG IS BOUND TO PLASMA ALBUMIN, LARGELY TO ALBUMIN. SMALL UNBOUND PORTION GAINS ACCESS TO GLOMERULAR FILTRATE; A MUCH LARGER PORTION IS ACTIVELY SECRETED BY PROXIMAL TUBULE. IN SPITE OF ITS LOW PKA (3.4), HIGH LIPID SOLUBILITY OF UNDISSOCIATED FORM RESULTS IN VIRTUALLY COMPLETE ABSORPTION BY BACK DIFFUSION UNLESS URINE IS MARKEDLY ALKALINE. SMALL AMOUNT OF PROBENECID GLUCURONIDE APPEARS IN URINE. ... /ORG ACID CMPD SUCH AS PROBENECID /ARE/ NOT TAKEN UP SO AVIDLY BY /PARENCHYMATOUS OR RETICULO-ENDOTHELIAL TISSUES/ & EXHIBIT HIGHER PLASMA CONCN ... . For more Absorption, Distribution and Excretion (Complete) data for PROBENECID (8 total), please visit the HSDB record page. Metabolism / Metabolites YIELDS P-DIPROPYLSULFAMOYLBENZOYL-BETA-D-GLUCURONIC ACID; P-(2-HYDROXYPROPYL N-PROPYLSULFAMOYL) BENZOIC ACID; P-(3-HYDROXYPROPYL N-PROPYLSULFAMOYL) BENZOIC ACID; & P-PROPYLSULFAMOYLBENZOIC ACID IN MAN. /FROM TABLE/ STRUCTURES OF ALL OF METAB OF PROBENECID IN RAT BILE & HUMAN URINE HAVE BEEN ELUCIDATED. PROPIONIC ACID HAS NOW BEEN IDENTIFIED AS ANOTHER PROBENECID METAB. MAJOR METABOLIC PATHWAYS INVOLVE SIDE-CHAIN OXIDATION & GLUCURONIDE CONJUGATION ... . ... BETA-GLUCURONIDES OF 2- & 3-HYDROXYLATED METAB & ACYL GLUCURONIDE OF PROBENECID PER SE HAVE NOW BEEN IDENTIFIED. ... THERE IS CONSIDERABLE SPECIES DIFFERENCE IN METABOLISM. IN RATS & MONKEYS OXIDATION IS FAVORED. ... IN DOGS CONJUGATION ... /IS/ MAJOR PATHWAY, WHEReAS IN MAN, OXIDATIVE ... PATHWAY ... IS AS IMPORTANT AS GLUCURONIDATION. CHRONIC ADMIN OF DRUGS NOT ONLY STIMULATES METAB OF OTHER CMPD, BUT IN SOME INSTANCES PHARMACOLOGICAL OR TOXIC EFFECT OF A DRUG WHEN GIVEN CHRONICALLY, DIMINISHES, BECAUSE IT STIMULATES ITS OWN METABOLISM EXAMPLE OF DRUG THAT EXERT THIS EFFECT IN DOGS ... /IS/ ... PROBENECID ... . For more Metabolism/Metabolites (Complete) data for PROBENECID (6 total), please visit the HSDB record page. Biological Half-Life 6-12 hours THE HALF-LIFE OF /PROBENECID/ IN PLASMA IS DOSE DEPENDENT AND VARIES FROM LESS THAN 5 HR TO MORE THAN 8 HR ... . Following oral administration of 2 g of probenecid, plasma half-life of the drug ranges from 4-17 hr; the half-life decreases as the dose decreases from 2 g to 500 mg. |
| Toxicity/Toxicokinetics |
23662399 man TDLo oral 630 mg/kg/6W KIDNEY, URETER, AND BLADDER: PROTEINURIS; KIDNEY, URETER, AND BLADDER: CHANGES PRIMARILY IN GLOMERULI Archives of Pathology., 94(241), 1972 [PMID:5051645] 23662399 mouse LD50 oral 1666 mg/kg BEHAVIORAL: CONVULSIONS OR EFFECT ON SEIZURE THRESHOLD; BEHAVIORAL: TETANY; LUNGS, THORAX, OR RESPIRATION: OTHER CHANGES Journal of Pharmacology and Experimental Therapeutics., 102(208), 1951 [PMID:14851208] 23662399 rat LD50 intraperitoneal 394 mg/kg BEHAVIORAL: CONVULSIONS OR EFFECT ON SEIZURE THRESHOLD; BEHAVIORAL: TETANY; LUNGS, THORAX, OR RESPIRATION: OTHER CHANGES Journal of Pharmacology and Experimental Therapeutics., 102(208), 1951 [PMID:14851208] 23662399 rabbit LD50 intravenous 304 mg/kg BEHAVIORAL: CONVULSIONS OR EFFECT ON SEIZURE THRESHOLD; BEHAVIORAL: TETANY; LUNGS, THORAX, OR RESPIRATION: OTHER CHANGES Journal of Pharmacology and Experimental Therapeutics., 102(208), 1951 [PMID:14851208] 23662399 mouse LD50 subcutaneous 1156 mg/kg BEHAVIORAL: CONVULSIONS OR EFFECT ON SEIZURE THRESHOLD; BEHAVIORAL: TETANY; LUNGS, THORAX, OR RESPIRATION: OTHER CHANGES Journal of Pharmacology and Experimental Therapeutics., 102(208), 1951 [PMID:14851208] Hepatotoxicity There are no reports on the frequency of liver test abnormalities during probenecid therapy, but they are probably rare as the drug is largely secreted unchanged in the urine. A single case report of a severe hypersensitivity reaction from probenecid and rechallenge with a rapid and severe recurrence of jaundice was reported over 50 years ago. As is typical for hypersensitivity reactions, the onset was within days of starting probenecid and was accompanied by fever and rash. Likelihood score: D (possible rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Limited information indicates that maternal doses of probenecid up to 2 grams daily produce low levels in milk and would not be expected to cause any adverse effects in breastfed infants, especially if the infant is older than 2 months. In animal studies, probenecid increased the breastmilk excretion of cimetidine, possible via an interaction with an active transport mechanism in the breast. The implications of enhanced excretion of drugs given with probenecid for nursing mothers and their infants has not been studied; however, only a few drugs are known to undergo active transport into breastmilk. ◉ Effects in Breastfed Infants A woman with mastitis received 3 days of intravenous cephalothin, followed by 16 days of probenecid 500 mg and cephalexin 500 mg 4 times daily for 16 days. Her infant developed green liquid stools, severe diarrhea, discomfort and crying. The authors judged the effects to be probably related to the cephalothin and cephalexin in milk rather than the probenecid. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding 75-95% |
| References |
[1]. Probenecid: novel use as a non-injurious positive inotrope acting via cardiac TRPV2 stimulation. J Mol Cell Cardiol. 2012 Jul;53(1):134-44. [2]. Interactions of the human multidrug resistance proteins MRP1 and MRP2 with organic anions. Mol Pharmacol. 2000 Apr;57(4):760-8. [3]. Probenecid inhibits the human bitter taste receptor TAS2R16 and suppresses bitter perception of salicin. PLoS One. 2011;6(5):e20123. [4]. Probenecid, a gout remedy, inhibits pannexin 1 channels. Am J Physiol Cell Physiol. 2008 Sep;295(3):C761-7. |
| Additional Infomation |
Probenecid appears as odorless white or almost white crystalline powder. Slightly bitter taste; pleasant aftertaste. (NTP, 1992) Probenecid is a sulfonamide in which the nitrogen of 4-sulfamoylbenzoic acid is substituted with two propyl groups. It has a role as a uricosuric drug. It is a sulfonamide and a member of benzoic acids. The prototypical uricosuric agent. It inhibits the renal excretion of organic anions and reduces tubular reabsorption of urate. Probenecid has also been used to treat patients with renal impairment, and, because it reduces the renal tubular excretion of other drugs, has been used as an adjunct to antibacterial therapy. Probenecid is a uricosuric agent used for the treatment of gout usually in combination with other agents. Probenecid has been associated with minor serum aminotransferase elevations and very rarely with hypersensitivity reactions which, even more rarely, can be accompanied by acute liver injury. Probenecid is a benzoic acid derivative with antihyperuricemic property. Probenecid competitively inhibits the active reabsorption of urate at the proximal tubule in the kidney thereby increasing urinary excretion of uric acid and lowering serum urate concentrations. This prevents urate deposition and promotes resolution of existing urate deposits. In addition, probenecid modulates the transport of organic acids and acidic drugs at the proximal and distal renal tubule, thereby increasing the drug serum concentration. The prototypical uricosuric agent. It inhibits the renal excretion of organic anions and reduces tubular reabsorption of urate. Probenecid has also been used to treat patients with renal impairment, and, because it reduces the renal tubular excretion of other drugs, has been used as an adjunct to antibacterial therapy. See also: Colchicine; probenecid (component of); Ampicillin/ampicillin trihydrate; probenecid (component of). Drug Indication For the reduction of serum uric acid concentrations in chronic gouty arthritis and tophaceous gout in patients with frequent disabling gout attacks. Has also been effectively used to promote uric acid excretion in hyperuricemia secondary to the administration of thiazide and related diuretics. Mechanism of Action Probenecid inhibits the tubular reabsorption of urate, thus increasing the urinary excretion of uric acid and decreasing serum urate levels. Probenecid may also reduce plasma binding of urate and inhibit renal secretion of uric acid at subtherapeutic concentrations. The mechanism by which probenecid inhibits renal tubular transport is not known, but the drug may inhibit transport enzymes that require a source of high energy phosphate bonds and/or nonspecifically interfere with substrate access to protein receptor sites on the kidney tubules. IN HIGHER DOSES THAN ARE REQUIRED FOR URICOSURIC EFFECT, PROBENECID ALSO INHIBITS TRANSPORT OF ORG ACIDS AT OTHER SITES, IE, TRANSPORT SYSTEM THAT REMOVES ORG ACIDS FROM CEREBROSPINAL FLUID. IT INHIBITS TUBULAR REABSORPTION OF URATE, THUS INCR URINARY EXCRETION OF URIC ACID & DECR SERUM URIC ACID LEVELS. Probenecid is a renal tubular blocking agent. The drug competitively inhibits active reabsorption of uric acid at the proximal convoluted tubule, thus promoting urinary excretion of uric acid and reducing serum urate concentrations. Probenecid may reduce plasma protein binding of urate and, in subtherapeutic doses, may inhibit renal secretion of uric acid. In healthy individuals, probenecid has no effect on the glomerular filtration rate or on the tubular reabsorption of normal urinary constituents such as glucose, arginine, urea, sodium, potassium, chloride, or phosphate. At the proximal and distal tubules, probenecid competitively inhibits the secretion of many weak organic acids including penicillins, most cephalosporins, and some other beta-lactam antibiotics. In general, the net effect of probenecid on the plasma concentration of weak acids depends on the ratio of the amount of organic acid secreted by the kidneys to that amount filtered at the glomeruli. Thus, probenecid substantially increases plasma concentrations of acidic drugs eliminated principally by renal secretion, but increases plasma concentrations only slightly if the drug is eliminated mainly by filtration. Plasma concentrations of penicillins are often more than doubled by probenecid; the concentration of penicillin in the CSF is also increased. Probenecid also substantially increases plasma concentrations of most cephalosporins and some other beta-lactam antibiotics. In addition, half-lives of the penicillins and cephalosporins are prolonged and their volumes of distribution may be reduced by probenecid. ... The cellular mechanism(s) responsible for the inhibition of renal tubular transport by probenecid is not known. The drug may inhibit transport enzymes that require a source of high energy phosphate bonds and/or nonspecifically interfere with substrate access to protein receptor sites on the kidney tubules. CSF concentrations of 5-hydroxyindoleacetic acid, homovanillic acid, cyclic adenosine monophosphate, and 4-hydroxy-3-methoxyphenylglycol are elevated following administration of probenecid. It has been proposed that probenecid blocks the active transport of these organic acids from the CSF into blood. Probenecid-induced elevations of homovanillic acid (a dopamine metabolite) in the CSF of patients with parkinsonian syndrome and of 5-hydroxyindoleacetic acid(a metabolite of serotonin) in the CSF of mentally depressed patients are substantially lower than those in healthy patients. |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2537 mL | 16.2686 mL | 32.5373 mL | |
| 5 mM | 0.6507 mL | 3.2537 mL | 6.5075 mL | |
| 10 mM | 0.3254 mL | 1.6269 mL | 3.2537 mL |