PTC-299 (PTC299; PTC 299) is a novel, potent and orally bioavailable VEGF inhibitor with potential antiangiogenesis and antineoplastic activities. It acts via posttranscriptional regulation of VEGF mRNA under conditions of cellular stress. PTC299 has broad and potent activity against hematological cancer cells. PTC299 targets post-transcriptionally by selectively binding the 5'- and 3'-untranslated regions (UTR) of VEGF messenger RNA (mRNA), thereby preventing translation of VEGF. This inhibits VEGF protein production and decreases its levels in the tumor and bloodstream. In turn, this may result in the inhibition of migration, proliferation and survival of endothelial cells, microvessel formation, the inhibition of tumor cell proliferation, and eventually the induction of tumor cell death.
Physicochemical Properties
| Molecular Formula | C25H20CL2N2O3 |
| Molecular Weight | 467.343904495239 |
| Exact Mass | 466.09 |
| Elemental Analysis | C, 64.25; H, 4.31; Cl, 15.17; N, 5.99; O, 10.27 |
| CAS # | 1256565-36-2 |
| PubChem CID | 49787172 |
| Appearance | White to light yellow solid powder |
| LogP | 6.3 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 3 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 32 |
| Complexity | 651 |
| Defined Atom Stereocenter Count | 1 |
| SMILES | COC1=CC=C(C=C1)[C@H]2C3=C(CCN2C(=O)OC4=CC=C(C=C4)Cl)C5=C(N3)C=CC(=C5)Cl |
| InChi Key | SRSHBZRURUNOSM-DEOSSOPVSA-N |
| InChi Code | InChI=1S/C25H20Cl2N2O3/c1-31-18-7-2-15(3-8-18)24-23-20(21-14-17(27)6-11-22(21)28-23)12-13-29(24)25(30)32-19-9-4-16(26)5-10-19/h2-11,14,24,28H,12-13H2,1H3/t24-/m0/s1 |
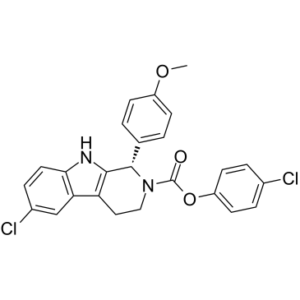
| Chemical Name | (4-chlorophenyl) (1S)-6-chloro-1-(4-methoxyphenyl)-1,3,4,9-tetrahydropyrido[3,4-b]indole-2-carboxylate |
| Synonyms | PTC299; PTC 299; PTC-299 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Dihydroorotate dehydrogenase (DHODH) (Inhibitor. IC50 ≈ 1 nM for inhibition of proliferation in MOLM-13 AML cells; binds to the same site as brequinar based on pull-down and docking studies) [1] |
| ln Vitro |
PTC299 suppresses the synthesis of VEGFA protein in HeLa cells induced by hypoxia, with an EC50 of 1.64 ± 0.83 nM[1]. In leukemia cells, PTC299 is more than ten to thousand times more effective than Brequinar, Vidofludimus, or A 77-1726, with an IC50 of about 1 nM [1]. PTC299 inhibited hypoxia-induced VEGFA protein production in HeLa cells with an EC50 of 1.64 ± 0.83 nM. The inactive R-enantiomer PTC-371 had no effect. [1] PTC299 dose-dependently inhibited the production of a V5-tagged VEGFA165 protein in HT1080 cells transfected with a construct containing the VEGFA 5'-UTR, confirming the requirement of this UTR for its activity. [1] Pulse-chase experiments using 35S-labeled methionine/cysteine showed that PTC299 (100 nM) decreased levels of newly synthesized VEGFA protein without affecting total protein synthesis, VEGFA secretion, or degradation, indicating inhibition of VEGFA mRNA translation. [1] PTC299 selectively decreased intracellular levels of pyrimidine nucleotides (dCTP, dTTP) but not purine nucleotides (dATP, dGTP) in HT1080 cells after 8-hour treatment. [1] Metabolic labeling with 15N-glutamine showed that PTC299 (100 nM) inhibited de novo pyrimidine nucleotide synthesis, reducing levels of 15N-labeled CTP, UTP, and UMP. This inhibition was rapid and dose-dependent. [1] The inhibition of VEGFA protein synthesis and S-phase cell cycle arrest induced by PTC299 could be completely rescued by the addition of exogenous uridine, but not by adenosine, cytidine, or guanosine, linking its effects to pyrimidine nucleotide depletion. [1] Chemical pull-down assays using PTC299-conjugated Sepharose beads specifically enriched DHODH and prohibitin-1/2 from cell lysates. Binding of DHODH, but not prohibitin, to the beads was competitively blocked by free PTC299 or brequinar. DHODH could also be eluted from the beads by free PTC299 or brequinar. PTC299 beads bound human DHODH but not rat DHODH. [1] PTC299 inhibited the activity of DHODH in isolated mitochondria from K562 cells with potency similar to brequinar and greater than teriflunomide. [1] In a panel of 240 tumor cell lines, PTC299 showed broad antiproliferative activity. Sensitivity (IC50 < 1 µM) was more frequent in hematopoietic lines (57%) than in solid tumor lines (18%). Gene expression analysis indicated that sensitive cells generally had lower expression of pyrimidine salvage enzymes (CDA, UPP1) and relied more on de novo synthesis. [1] PTC299 potently inhibited the proliferation of leukemia cell lines (e.g., MOLM-13 AML, IC50 ≈ 1 nM) and was significantly more potent than brequinar, vidofludimus, or teriflunomide. It also inhibited proliferation of five patient-derived acute myeloid leukemia (AML) cell samples with IC50 values ranging from 2 nM to 60 nM. [1] PTC299 induced differentiation in AML MOLM-13 cells. [1] |
| ln Vivo |
In a mouse HT1080 fibrosarcoma xenograft model, oral administration of PTC299 (0.3, 1, 3 mg/kg, twice daily) dose-dependently inhibited tumor growth. The 3 mg/kg BID dose achieved maximal effect. [1] In the same model, PTC299 treatment caused a dose-dependent reduction in intratumor and circulating plasma human VEGFA levels, correlating with tumor growth control. Tumor vessel diameter was reduced and distribution was more uniform as shown by CD31 staining. [1] In a systemic leukemia model, NOD-SCID mice intravenously inoculated with MOLT-4 human acute lymphoblastic leukemia (ALL) cells were treated with PTC299 (10 mg/kg, once daily) starting 7 days post-inoculation. PTC299 significantly prolonged median survival time (136 days vs 46 days for vehicle) and reduced circulating leukemia cells. Doxorubicin did not prolong survival. [1] In a solid tumor xenograft model, nude mice subcutaneously implanted with MOLM-13 AML cells were treated with PTC299 (10 mg/kg, QD), cytarabine (AraC, 60 mg/kg, QD), or their combination. PTC299 alone significantly delayed tumor growth compared to vehicle or AraC alone. The combination of PTC299 and AraC further delayed tumor growth. [1] In a clinical study of patients with neurofibromatosis type 2 (NF2), 8 weeks of PTC299 treatment significantly decreased elevated serum VEGFA levels. Analysis of archived serum samples from some patients showed that PTC299 treatment increased levels of dihydroorotate (DHO), the substrate of DHODH, indicating target engagement in humans. [1] |
| Enzyme Assay |
For the in vitro DHODH activity assay, a chromogen reduction assay was used. The oxidation of dihydroorotic acid (DHO) coupled to the reduction of ubiquinone was measured by monitoring the reduction of 2,6-dichlorophenolindophenol (DCIP) spectrophotometrically. The reaction velocity was determined in the presence of test compounds or vehicle control. [1] For DHODH inhibition studies using a more physiological context, mitochondria were isolated from K562 cells in log-phase growth using a Dounce homogenizer. The isolated mitochondria were pelleted and resuspended in homogenization buffer. DHODH activity in these mitochondrial preparations was then assessed in the presence of inhibitors using the same chromogenic assay principle, with orotate production quantified by LC-MS/MS after a 30-minute reaction. [1] |
| Cell Assay |
For the VEGFA inhibition ELISA, cells (e.g., HeLa, HT1080) were treated with a series of doses of PTC299 for a specified time (e.g., under hypoxic conditions for HeLa). The culture supernatant was then collected, and VEGFA protein levels were quantified using a commercial enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer's instructions. Dose-response curves were generated, and EC50 values were calculated. [1] For cell proliferation/cytotoxicity assays (CC50 or IC50 determination), cells were seeded in plates and treated with a series of concentrations of PTC299 for 72 hours. Cell viability was then assessed using the CellTiter-Glo Luminescent Cell Viability Assay, which measures cellular ATP levels as an indicator of viable cell number. Dose-response curves were plotted, and IC50 values were calculated. [1] For metabolic labeling of de novo pyrimidine synthesis, cells in log-phase growth were cultured in glutamine-free medium containing 1 mM 15N-glutamine. After treatment with compounds or vehicle for specified times (e.g., 8 hours), cells were washed, harvested, and lysed in a cold methanol/water mixture. The supernatant was collected after centrifugation and analyzed by LC-MS/MS to quantify 15N-labeled pyrimidine nucleotide metabolites (e.g., UMP, CTP, UTP). [1] For cell cycle analysis, HT1080 cells were treated with PTC299 (100 nM) in the presence or absence of exogenous nucleosides (100 µM) for 24 hours. Cells were then harvested, fixed, stained with propidium iodide, and analyzed by flow cytometry to determine DNA content and cell cycle distribution. [1] |
| Animal Protocol |
For subcutaneous xenograft models (e.g., HT1080 fibrosarcoma, MOLM-13 AML), tumor cells were implanted into the flank of immunodeficient mice (e.g., nude mice). When tumors reached a predetermined size, mice were randomized into treatment groups. PTC299 was formulated in a vehicle (specific composition not detailed in the main text) and administered orally by gavage. In the HT1080 study, doses of 0.3, 1, and 3 mg/kg were given twice daily (BID). In the MOLM-13 AML study, a dose of 10 mg/kg was given once daily (QD). Tumor volumes were measured regularly. [1] For the systemic leukemia model, NOD-SCID mice were inoculated intravenously with 1 × 107 MOLT-4 human ALL cells. Seven days post-inoculation, mice were randomized and treatment began. PTC299 was administered orally at 10 mg/kg once daily (QD). Control groups received vehicle or doxorubicin (0.5 or 1 mg/kg, intraperitoneally, once per week). Mice were monitored for survival. Circulating leukemia cells were quantified at weeks 2 and 4 by flow cytometry staining for human CD45. [1] |
| ADME/Pharmacokinetics |
Analysis of archived serum samples from a patient in the NF2 clinical trial showed that plasma concentrations of PTC299 increased with treatment and were correlated with increased serum dihydroorotate (DHO) levels, confirming systemic exposure and target engagement. [1] |
| Toxicity/Toxicokinetics |
Preclinical safety studies (referenced in supplementary materials) indicated that PTC299 was safe and well-tolerated in animal models. It did not inhibit the proliferation of normal primary cells, including hematopoietic stem cells, at concentrations 1000-fold greater than its IC50 in tumor cells. [1] PTC299 showed no off-target inhibition against a panel of 205 kinases and 62 other pharmacological targets in screening assays. [1] In previous clinical trials for solid tumors, PTC299 did not cause myeloid suppression, a side effect associated with some other DHODH inhibitors. [1] However, the literature notes that PTC299 was placed on clinical hold following the occurrence of severe hepatotoxicity in two out of 279 treated individuals in prior clinical studies. [1] |
| References |
[1]. Targeting of Hematologic Malignancies with PTC299, A Novel Potent Inhibitor of DihydroorotateDehydrogenase with Favorable Pharmaceutical Properties. Mol Cancer Ther. 2019 Jan;18(1):3-16. [2]. Brief Report: A Phase 1b/Pharmacokinetic Trial of PTC299, a Novel PostTranscriptional VEGF Inhibitor, for AIDS-Related Kaposi's Sarcoma: AIDS Malignancy Consortium Trial 059. J Acquir Immune Defic Syndr. 2016 May 1;72(1):52-7. [3]. Phase I and pharmacokinetic trial of PTC299 in pediatric patients with refractory or recurrent central nervous system tumors: a PBTC study. J Neurooncol. 2015;121(1):217-224. [4]. Zeng Z, et al, Targeting dihydroorotate dehydrogenase in acute myeloid leukemia. Haematologica. 2018 Sep;103(9):1415-1417. |
| Additional Infomation |
PTC299 is a novel, orally administered small-molecule designed to inhibit the production of vascular endothelial growth factor (VEGF) in tumors. Overexpression of VEGF plays a key role in multiple diseases including cancer and macular degeneration. PTC299 was discovered through PTC's GEMS technology by targeting the post-transcriptional processes that regulate VEGF formation, and is currently being developed for the treatment of cancer. Emvododstat is an orally bioavailable, small molecule inhibitor of vascular endothelial growth factor (VEGF) synthesis with potential antiangiogenesis and antineoplastic activities. Emvododstat targets post-transcriptionally by selectively binding the 5'- and 3'-untranslated regions (UTR) of VEGF messenger RNA (mRNA), thereby preventing translation of VEGF. This inhibits VEGF protein production and decreases its levels in the tumor and bloodstream. In turn, this may result in the inhibition of migration, proliferation and survival of endothelial cells, microvessel formation, the inhibition of tumor cell proliferation, and eventually the induction of tumor cell death. VEGFs are upregulated in a variety of tumor cell types and play key roles during angiogenesis. In addition, emvododstat may enhance the antitumor activity of other chemotherapeutic agents. Drug Indication Investigated for use/treatment in cancer/tumors (unspecified) and solid tumors. Mechanism of Action PTC299 was designed to inhibit VEGF production in tumors by targeting the post-transcriptional control processes that regulate VEGF formation. Because PTC299 inhibits VEGF production, its action occurs at a different point in the VEGF pathway than therapies, such as Avastin® or Sutent®. PTC299 may be active both as a single agent or when used in combination with other anti-angiogenic agents or with chemotherapy agents for the treatment of cancers. Pharmacodynamics PTC299 demonstrated a broad range of activity in blocking VEGF synthesis in multiple tumor types, including breast, cervical, colorectal, fibrosarcoma, gastric, lung, melanoma, neuroblastoma, ovarian, pancreatic, prostate and renal cell cancer lines. PTC299 as a monotherapy significantly reduced VEGF concentrations in tumors and plasma, reduced tumor blood vessel density, and substantially impeded tumor progression. PTC299 was initially identified from a phenotypic screen as an inhibitor of VEGFA mRNA translation via PTC Therapeutics' GEMS technology. Subsequent mechanistic studies revealed that its primary target is dihydroorotate dehydrogenase (DHODH), a rate-limiting enzyme in the de novo pyrimidine biosynthesis pathway. Inhibition of VEGFA production is a downstream consequence of pyrimidine nucleotide depletion. [1] It represents a novel class of DHODH inhibitors. Unlike some inhibitors identified using purified enzyme assays, PTC299 shows superior potency when DHODH is embedded in the mitochondrial membrane, suggesting its activity may be facilitated by membrane lipid interactions. [1] Docking studies indicate that PTC299 binds to the same hydrophobic pocket in DHODH as the brequinar analog C44, mainly through non-polar lipophilic ("hand-in-glove") interactions. [1] Its activity is species-selective, potently inhibiting human DHODH but not rat DHODH. [1] Due to its potent activity against hematologic cancer cells in vitro and in vivo, and the observation that such cancers often have reduced pyrimidine salvage pathway activity, PTC299 is being re-evaluated for the treatment of hematologic malignancies (e.g., leukemia, lymphoma). [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~50 mg/mL (~106.99 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 0.83 mg/mL (1.78 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 8.3 mg/mL clear DMSO stock solution to 400 μL of PEG300 and mix evenly; then add 50 μL of Tween-80 to the above solution and mix evenly; then add 450 μL of normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 0.83 mg/mL (1.78 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 8.3 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1398 mL | 10.6988 mL | 21.3977 mL | |
| 5 mM | 0.4280 mL | 2.1398 mL | 4.2795 mL | |
| 10 mM | 0.2140 mL | 1.0699 mL | 2.1398 mL |