PQR-530 is a novel, potent, orally bioavailable and brain-penetrant dual pan-PI3K/mTORC1/2 inhibitor, it exhibits antitumor activity. PQR530 inhibited protein kinase B (PKB, pSer473) and ribosomal protein S6 (pS6, pSer235/236) phosphorylation with IC50 values of 0.07 µM. PQR530 showed excellent selectivity over a wide panel of kinases, as well as excellent selectivity versus unrelated receptor enzymes and ion channels. Moreover, PQR530 displayed potency in a panel of 44 cancer cell lines (NTRC OncolinesTM) to prevent cancer cell growth (mean value for GI50 of 426 nM). Oral application of PQR530 to mice resulted in a dose-proportional PK and demonstrated good oral bioavailability and excellent brain penetration.
Physicochemical Properties
| Molecular Formula | C18H23F2N7O2 |
| Molecular Weight | 407.417729616165 |
| Exact Mass | 407.188 |
| CAS # | 1927857-61-1 |
| PubChem CID | 121334433 |
| Appearance | Light yellow to yellow solid powder |
| Density | 1.338±0.06 g/cm3 |
| LogP | 1.4 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 11 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 29 |
| Complexity | 528 |
| Defined Atom Stereocenter Count | 1 |
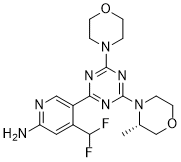
| SMILES | C[C@H]1COCCN1C2=NC(=NC(=N2)N3CCOCC3)C4=CN=C(C=C4C(F)F)N |
| InChi Key | SYKBZXMKAPICSO-NSHDSACASA-N |
| InChi Code | InChI=1S/C18H23F2N7O2/c1-11-10-29-7-4-27(11)18-24-16(13-9-22-14(21)8-12(13)15(19)20)23-17(25-18)26-2-5-28-6-3-26/h8-9,11,15H,2-7,10H2,1H3,(H2,21,22)/t11-/m0/s1 |
| Chemical Name | (S)-4-(difluoromethyl)-5-(4-(3-methylmorpholino)-6-morpholino-1,3,5-triazin-2-yl)pyridin-2-amine |
| Synonyms | PQR-530; PQR-0530; PQR 530; PQR 0530; PQR530; PQR0530 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Dual pan-class I PI3K and mTOR kinase inhibitor. (Kd for PI3Kα = 0.84 nM, mTOR = 0.33 nM; Ki for PI3Kα = 11.1 nM, mTOR = 7.4 nM). [2] |
| ln Vitro |
Strong oral dual pan-PI3K/mTORC1/2 inhibitor with anti-tumor action, PQR-530 penetrates the brain. PQR-530 inhibits mTOR complex C1/2 and all PI3K isoforms with great potency and selectivity. With an IC50 value of 0.07 µM, PQR-530 suppresses the phosphorylation of ribosomal protein S6 (pS6, pSer235/236) and protein kinase B (PKB, pSer473) in A2058 melanoma cells. With an average GI50 of 426 nM, PQR-530 has inhibitory efficacy against the development of 44 cancer cell lines[1]. PQR530 potently inhibited cellular PI3K/mTOR signaling in A2058 melanoma cells, with IC50 values of 62.2 nM for phosphorylation of PKB/Akt (Ser473) and 61.9 nM for phosphorylation of ribosomal protein S6 (Ser235/236). [2] In a panel of 66 cancer cell lines, PQR530 exhibited a mean half-maximal growth inhibition (GI50) of 0.43 µM. The compound acted primarily cytostatically, with cytotoxicity (LD50) observed in only about 10% of cell lines at concentrations well above the GI50. [2] The cellular sensitivity profiles of PQR530 and the dual PI3K/mTOR inhibitor GDC-0980 (3) across 44 tumor cell lines showed good overall correlation. [2] Compared to the pan-PI3K inhibitor PQR309 (5), PQR530 showed increased potency (2.5- to 3-fold lower average GI50 and IC50) in growth inhibition across cell lines, attributed to its enhanced mTOR inhibition. [2] In A2058 and SKOV3 cells, PQR530 effectively suppressed phosphorylation of downstream targets including PKB/Akt, GSK3β, p70S6K, and S6, confirming its dual pathway blockade. [2] |
| ln Vivo |
In an OVCAR-3 human ovarian cancer xenograft model in BALB/c nude mice, daily oral administration of PQR530 (25 mg/kg, QD) starting from day 17 post-inoculation significantly inhibited tumor growth compared to the vehicle control group. No significant body weight loss was observed in the treatment group. [2] |
| Enzyme Assay |
The binding affinity (Ki) of compounds for PI3Kα and mTOR was determined using a time-resolved Förster resonance energy transfer (TR-FRET) assay. For PI3Kα, a His-tagged recombinant protein was used with a biotinylated anti-His antibody and europium-labeled streptavidin. The tracer was an AlexaFluor647-labeled kinase tracer. The assay buffer contained HEPES, MgCl2, EGTA, and a detergent. [2] For mTOR, a truncated GST-fusion protein was used with a europium-labeled anti-GST antibody. The assay buffer contained HEPES, MgCl2, EGTA, and Pluronic F-127. Inhibitor Ki values were calculated from dose-response curves. [2] The selectivity profile over a wide range of kinases was assessed using a commercial kinase binding panel (KINOMEScan). Compounds were tested at a single concentration (10 µM) or in dose-response to determine binding constants. Selectivity scores S(35) and S(10) were calculated based on the fraction of kinases with activity reduced below 35% or 10% of control, respectively. [2] |
| Cell Assay |
Cellular potency for inhibiting PI3K/mTOR signaling was analyzed using an In-Cell Western assay. Cells (e.g., A2058, SKOV3) were seeded in 96-well plates, treated with inhibitors for 1 hour, then fixed. Phosphorylation of PKB/Akt on Ser473 and ribosomal S6 on Ser235/236 was detected using specific primary antibodies. Cellular tubulin was used as a loading control, detected with a mouse anti-α-tubulin antibody. Fluorescent secondary antibodies were used, and signals were quantified using an infrared imaging scanner. The percentage of remaining phospho-substrate signal was normalized to tubulin. [2] Cell proliferation assays were performed by seeding cells in 96-well plates 24 hours before treatment. Inhibitors were added at indicated concentrations for 72 hours. Cells were then fixed, stained with Hoechst 33324, and nuclei were counted using a high-content imaging system. Dose-response curves were generated to calculate GI50 and LD50 values. [2] For a larger cell panel (66 cancer cell lines), cells were exposed to serial dilutions of PQR530 for 72 hours, and viability was assessed using a luminescence-based assay. GI50 and LD50 values were calculated by nonlinear regression fitting of the dose-response data. [2] |
| Animal Protocol |
For pharmacokinetic studies in male C57BL/6J mice, PQR530 was formulated by first dissolving in DMSO, then adding an aqueous solution of hydroxypropyl-β-cyclodextrin to achieve the final dosing solution. A single oral dose of 50 mg/kg was administered. Blood plasma, brain, and thigh muscle tissues were collected at various time points. Drug concentrations were quantified using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Pharmacokinetic parameters (Cmax, Tmax, t1/2, AUC, Cl) were calculated. [2] For the OVCAR-3 xenograft efficacy study, female BALB/c nude mice were inoculated subcutaneously with tumor cells. When the mean tumor volume reached approximately 157 mm³ (day 17), mice were randomized into groups. The treatment group received PQR530 at 25 mg/kg orally, once daily (QD). The control group received vehicle. Body weight and tumor dimensions (measured by caliper) were monitored multiple times per week. Tumor volume was calculated. The study was terminated on day 45, and excised tumors were weighed. [2] |
| ADME/Pharmacokinetics |
In vitro metabolic stability of PQR530 was high in liver microsomes from human, rat, dog, and mouse, with >73% compound remaining after 30 minutes. [2] In hepatocyte cultures from the same species, intrinsic clearance was low, and half-lives ranged from 142 to 250 minutes. The half-life in human hepatocytes was approximately 4.2 hours. [2] After a single oral dose (50 mg/kg) in male C57BL/6J mice, PQR530 showed good oral bioavailability. Maximum concentration (Cmax) was reached at 0.5 hours (Tmax) in plasma, brain, and muscle. The plasma half-life (t1/2) was about 4.7 hours. [2] Brain penetration was excellent, with brain concentrations exceeding plasma levels (brain/plasma distribution ratio ~1.67 based on Cmax and AUC). [2] Permeability assays (PAMPA, MDCK) indicated fast passive diffusion. The compound was not a substrate for P-glycoprotein transport. [2] The fraction unbound (fu) in brain tissue binding assay was approximately 8%. [2] |
| Toxicity/Toxicokinetics |
In a broad off-target panel (CEREP BioPrint) at 10 µM, PQR530 showed only very weak competition for receptors, channels, transporters, and enzymes, suggesting a clean safety profile. [2] Initial toxicity studies in rats and dogs established no-observed-adverse-effect levels (NOAEL). In Wistar rats, NOAEL was 1 mg/kg/day for males and 0.5 mg/kg/day for females. In Beagle dogs, NOAEL was 1 mg/kg/day for males and 3 mg/kg/day for females. [2] Side effects observed were consistent with the mechanism of action (PI3K/mTOR inhibition), including transient increases in plasma glucose and insulin levels in mice after dosing. [2] |
| References |
[1]. Abstract 140: Discovery and biological evaluation of PQR530, a highly potent dual pan-PI3K/mTORC1/2 inhibitor. Cancer Res 2017;77(13 Suppl). [2]. (S)-4-(Difluoromethyl)-5-(4-(3-methylmorpholino)-6-morpholino-1,3,5-triazin-2-yl)pyridin-2-amine (PQR530), a Potent, Orally Bioavailable, and Brain-Penetrable Dual Inhibitor of Class I PI3K and mTOR Kinase. J Med Chem. 2019;62(13):6241-62. |
| Additional Infomation |
PQR530 is a brain-penetrant, ATP-competitive dual inhibitor developed from a structure-activity relationship (SAR) study based on PQR309. It incorporates an (S)-3-methylmorpholine group and a 4-difluoromethylpyridine moiety, which contributed to increased mTOR affinity and optimal physicochemical properties for brain penetration. [2] The binding mode was elucidated by molecular modeling and X-ray crystallography (PDB ID: 6OAC), confirming interactions within the ATP-binding pocket of PI3Kα. [2] The compound is proposed as a clinical candidate for oncology, particularly for cancers where brain penetration is desirable. [2] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~33.33 mg/mL (~81.81 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: 2.5 mg/mL (6.14 mM) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (6.14 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (6.14 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4545 mL | 12.2723 mL | 24.5447 mL | |
| 5 mM | 0.4909 mL | 2.4545 mL | 4.9089 mL | |
| 10 mM | 0.2454 mL | 1.2272 mL | 2.4545 mL |