PLX7904 (also known as PLX-7904; PB04; PB-04) is a novel potent and selective paradox-breaker B-Raf inhibitor with anticancer activity. It is also known as PLX-7904, PB04, and PB-04. In cells expressing mutant RAS, it blocks BRAFV600E with an IC50 value of less than 5 nM. Its IC50 values are 0.17 μM, 0.53 μM, and 0.16 μM for the two melanoma cell lines (A375 and COLO829) as well as the human colorectal cancer cell line COLO205 that expressed BRAFV600E, respectively. In mutant BRAF melanoma cells, ERK1/2 activation can be effectively inhibited by PLX7904, but in mutant RAS-expressing cells, ERK1/2 is not overexpressed. In mutant BRAF melanoma cells, ERK1/2 activation can be effectively inhibited by PLX7940, but in mutant RAS-expressing cells, ERK1/2 is not overexpressed.
Physicochemical Properties
| Molecular Formula | C24H22F2N6O3S | |
| Molecular Weight | 512.53 | |
| Exact Mass | 512.144 | |
| Elemental Analysis | C, 56.24; H, 4.33; F, 7.41; N, 16.40; O, 9.36; S, 6.26 | |
| CAS # | 1393465-84-3 | |
| Related CAS # |
|
|
| PubChem CID | 90116945 | |
| Appearance | White to light brown solid powder | |
| Density | 1.5±0.1 g/cm3 | |
| Index of Refraction | 1.672 | |
| LogP | 1.58 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 10 | |
| Rotatable Bond Count | 8 | |
| Heavy Atom Count | 36 | |
| Complexity | 895 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | S(N([H])C1C([H])=C([H])C(=C(C=1F)C(C1=C([H])N([H])C2=C1C([H])=C(C([H])=N2)C1C([H])=NC(C2([H])C([H])([H])C2([H])[H])=NC=1[H])=O)F)(N(C([H])([H])[H])C([H])([H])C([H])([H])[H])(=O)=O |
|
| InChi Key | DKNZQPXIIHLUHU-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C24H22F2N6O3S/c1-3-32(2)36(34,35)31-19-7-6-18(25)20(21(19)26)22(33)17-12-30-24-16(17)8-14(9-29-24)15-10-27-23(28-11-15)13-4-5-13/h6-13,31H,3-5H2,1-2H3,(H,29,30) | |
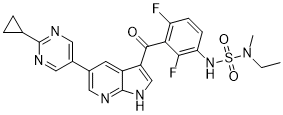
| Chemical Name | 5-(2-cyclopropylpyrimidin-5-yl)-3-[3-[[ethyl(methyl)sulfamoyl]amino]-2,6-difluorobenzoyl]-1H-pyrrolo[2,3-b]pyridine | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
BRaf(V600E) (IC50 = 5 nM)
BRAF V600E (IC50 = 14 nM), CRAF (IC50 = 4 nM), BRAF WT (IC50 = 31 nM), ARAF (IC50 = 10 nM) [1] - BRAF V600E (IC50 = 12 nM), CRAF (IC50 = 5 nM), BRAF WT (IC50 = 28 nM), ARAF (IC50 = 11 nM) [3] |
| ln Vitro |
PLX7940 is able to efficiently inhibit activation of ERK1/2 in mutant BRAF melanoma cells but does not hyperactivate ERK1/2 in mutant RAS-expressing cells. In mutant N-RAS mediated vemurafenib-resistant cells, PLX7904 promotes apoptosis and inhibits entry into the S phase as well as anchorage-independent growth, which is consistent with ERK1/2 re-activation driving the re-acquisition of malignant properties. Additionally, PLX7904 is being tested in human SCC cell line A431 and human breast adenocarcinoma cell line SKBR3 because these cells activate the MAPK pathway by upstream signals feeding into RAS (through overexpression of the EGFR and HER2 receptors, respectively)[1][2]. Treatment with PLX7904 inhibited ERK1/2 phosphorylation in BRAF V600E mutant melanoma cells (A375, SK-MEL-28) at concentrations ≥ 100 nM, with complete inhibition observed at 1 μM. The drug showed antiproliferative activity against these cells with GI50 values of 45 nM (A375) and 62 nM (SK-MEL-28). No paradoxical ERK activation was detected in BRAF WT cells (HEK293, MCF-7) even at concentrations up to 10 μM [1] - In vemurafenib-resistant melanoma cells harboring NRAS Q61K mutation (WM1366), PLX7904 reduced ERK1/2 phosphorylation in a dose-dependent manner, with significant inhibition at 500 nM. The compound exhibited antiproliferative effects with a GI50 of 320 nM and induced apoptosis in these cells, as evidenced by increased caspase-3/7 activity [3] - In BRAF V600E splice variant (BRAF V600E ΔEx3-8) expressing cells, PLX7904 inhibited ERK phosphorylation and cell proliferation with a GI50 of 58 nM, outperforming vemurafenib (GI50 = 420 nM) [2] |
| ln Vivo |
PLX7904 inhibits the COLO205 xenograft growth in eight mice per group[1].
When tested in vivo, subcutaneous B9-tumour growth was accelerated by vemurafenib but not by the equally potent BRAFV600E inhibitor PLX7904 when administered at the same dose.[1] PLX7904 administered orally at 50 mg/kg twice daily for 14 days significantly inhibited tumor growth in A375 (BRAF V600E) xenograft mice, achieving a tumor growth inhibition (TGI) rate of 89%. Tumor lysates showed reduced ERK1/2 phosphorylation and Ki67 expression, indicating suppressed proliferation [1] - In WM1366 (NRAS Q61K) xenograft model, oral administration of PLX7904 at 100 mg/kg twice daily for 21 days resulted in a TGI of 76%, with no significant weight loss or overt toxicity observed in mice [3] - In mice bearing BRAF V600E ΔEx3-8 mutant xenografts, PLX7904 (75 mg/kg, oral, twice daily) induced 82% TGI, whereas vemurafenib (100 mg/kg) only achieved 35% TGI [2] |
| Enzyme Assay |
PLX7904 (also known as PB04) is a novel potent and selective paradox-breaker B-Raf inhibitor with IC50 of ~5 nM against BRAFV600E in mutant RAS expressing cells. Biochemical assays and kinome selectivity profiling. [1] The in vitro RAF kinase activities were determined by measuring phosphorylation of a biotinylated substrate peptide as described previously25. PLX7904 was also tested against a panel of 287 kinases at concentrations of 1 μM in duplicate. Kinases inhibited by over 50% were followed up by IC50 determination. The 287 kinases represent all major branches of the kinome phylogenetic tree. The inhibition screen of 287 kinases was performed under contract as complementary panels at CRO companies. Phospho-ERK AlphaScreen assay. [1] To determine the effects of compound treatment on phosphorylation of ERK1/2, cells were plated in a 96-well plate and treated with an eight-point titration of compound for 1 h at 37 °C before lysis. To detect pERK, cell lysates were incubated with streptavidin-coated AlphaScreen donor beads, anti-mouse IgG AlphaScreen acceptor beads, a biotinylated anti-ERK1/2 rabbit antibody, and a mouse antibody that recognized ERK1/2 only when it was phosphorylated on Thr202 and Tyr204. The biotinylated ERK1/2 antibody bound both to the streptavidin-coated AlphaScreen donor beads and to ERK1/2 (regardless of its phosphorylation state), and the phospho-ERK1/2 antibody bound to the acceptor beads and to ERK1/2 that was phosphorylated at Thr202/Tyr204. An increase in ERK1/2 phosphorylation at Thr202/Tyr204 brought the donor and acceptor AlphaScreen beads into close proximity, generating a signal that could be quantified on an EnVision reader. Inhibition of ERK phosphorylation resulted in a loss of signal compared with DMSO controls. Kinase activity assays were performed using recombinant BRAF V600E, CRAF, BRAF WT, and ARAF proteins. Compounds were serially diluted and incubated with kinases and ATP for 60 minutes at 30°C. Phosphorylated substrates were detected via luminescence, and IC50 values were calculated from dose-response curves [1] - For BRAF splice variant enzyme activity measurement, recombinant BRAF V600E ΔEx3-8 protein was used. The assay protocol included a 45-minute incubation of the enzyme with PLX7904 and ATP, followed by substrate phosphorylation detection using a fluorescent readout [2] - NRAS mutant-associated kinase activity was assessed by incubating recombinant CRAF (activated by NRAS Q61K) with PLX7904 for 50 minutes at 37°C. Phosphorylation of MEK1 was quantified via ELISA to determine inhibitory potency [3] |
| Cell Assay |
For MTT assays, 2×103 cells are seeded in triplicate in 96 wells of their regular culture medium (which contains PLX4720 for PRT lines). The medium is replaced the following day with the specified RAF inhibitor after cells have been washed twice with PBS. The medium is changed after 48 hours, and then after an additional 48 hours, 10 μL of the 5 mg/mL MTT reagent is added to the wells and incubated for three hours. Then, formazan crystals are solubilized over night with a 1:10 dilution of 0.1 M glycine (pH 10.5) in DMSO. Then, using a Multiskan® Spectrum spectrophotometer, wells are evaluated at 450 nM. The results shown are a composite of three separate experiments and have been normalized to DMSO conditions. The error bars that are displayed are an accurate representation of the standard error of the mean. Anchorage-independent growth assay. [1] Twenty-five thousand B9 cells were plated in each well of a six-well plate with a bottom layer of 1% and a top layer of 0.4% low melting agar containing RPMI1640 medium with 10% FBS. For the RAF inhibitor study, B9 cells grown in soft agar were treated with vemurafenib, PLX4720 or PLX7904 at the indicated concentrations, or DMSO at 0.2% final concentration for 3 weeks. For the EGFR ligand study, B9 cells grown in soft agar were treated with AREG, TGF-α , or HB-EGF at the indicated concentrations for 3 weeks. For the vemurafinib and erlotinib combination study, B9 cells grown in soft agar were treated with vemurafenib, erlotinib, or a combination of the two compounds at the indicated concentrations, or DMSO for 3 weeks. Anchorage-independent colonies ≥ 100 μm were scored using AxioVision Rel 4.8 software. [1] Microarray gene expression analysis. [1] B9 cells were plated in 1 µM vemurafenib, 1 µM PLX7904 or 0.2% DMSO vehicle control and incubated for 17 h. Cells were harvested, total RNA was isolated, and gene expression was measured using Affymetrix Mouse420_2 chips following the manufacturer’s instructions. Vemurafenib response genes were identified by requiring the ratio between the treated and vehicle control samples be more than 1.9 (upregulated) or less than 0.54 (downregulated). [1] Antiproliferation assay: Cells were seeded in 96-well plates and treated with serial dilutions of PLX7904 for 72 hours. Cell viability was measured using a colorimetric assay, and GI50 values were calculated. Western blot analysis was performed on cell lysates to detect ERK1/2, phospho-ERK1/2, and caspase-3 levels [1] - For vemurafenib-resistant cells, cells were pre-treated with vemurafenib (1 μM) for 7 days to confirm resistance, then treated with PLX7904. Apoptosis was assessed by flow cytometry using Annexin V/PI staining, and clone formation assays were performed by seeding treated cells in 6-well plates and counting colonies after 14 days [3] - BRAF splice variant-expressing cells were treated with PLX7904 or vemurafenib for 48 hours. RNA was extracted for PCR analysis of BRAF splice variant expression, and immunofluorescence was used to visualize phospho-ERK localization [2] |
| Animal Protocol |
Bovine insulin is added to DMEM with 10% FBS, 1% penicillin/streptomycin, and 1% bovine insulin during the culture of COLO205 tumor cells at 37°C. Female Balb/C nude mice, 6–8 weeks old, weighing about 18–22 g, are subcutaneously injected at the right flank with COLO205 tumor cells (5×106) in 0.1 mL of PBS mixed with matrigel (50:50) to test the development of tumors. Eight mice are randomly assigned to each treatment group so that the mean weight and tumor size are balanced when the mean tumor size reaches about 100 mm3. DMEM 10% FBS 1% penicillin/streptomycin is used to grow B9 cells. The cells are trypsinized, washed three times with 20 mL RPMI, and then re-suspended, counted, and volume-adjusted to a final concentration of 5×107 cells per milliliter before final centrifugation. In 6- to 7-week-old female nude Balb/c mice, 5×106 cells are subcutaneously injected to begin B9 xenografts. When the average tumor size reaches 50–70 mm3, compound dosing begins. To maintain a balance between the average tumor size and body weight, animals are evenly distributed among treatment groups (n=10). Animals are given vehicle, vemurafenib 50 mg per kg, or PLX7904 50 mg per kg twice daily for days 1 through 14 and once daily for days 15 through 28. In weeks three and four, 2 g of 12-O-tetradecanoylphorbol-13-acetate (TPA) in 200 l of acetone is applied to the skin of every mouse. COLO205 tumour cells were cultured in DMEM 10% FBS 1% penicillin/streptomycin supplemented with bovine insulin, at 37 °C. Balb/C nude mice, female, 6–8 weeks old, weighing approximately 18–22 g, were inoculated subcutaneously at the right flank with COLO205 tumour cells (5 × 106) in 0.1 ml of PBS mixed with matrigel (50:50) for tumour development. The treatment was started when mean tumour size reached approximately 100 mm3, with eight mice in each treatment group randomized to balance the average weight and tumour size. B9 cells were expanded in DMEM 10% FBS 1% penicillin/streptomycin. Upon trypsinization the cells were washed three times with 20 ml RPMI, and after the final centrifugation were re-suspended, counted, and adjusted by volume to a final concentration of 5 × 107 cells per millilitre. B9 xenografts were started by injection of 5 × 106 cells subcutaneously in 6- to 7-week-old female nude Balb/c mice. Compound dosing started when the average size of tumours reached 50–70 mm3. Animals were equally distributed over treatment groups (n = 10) to balance the average tumour size and body weight. Animals were dosed orally for days 1–14 twice daily and days 15–28 once daily with vehicle, vemurafenib 50 mg per kg, or PLX7904 50 mg per kg. 12-O-tetradecanoylphorbol-13-acetate (TPA) was put on the skin of all mice twice a week during weeks 3 and 4 at a dose of 2 µg in 200 µl acetone. [1] Xenograft model establishment: Female nude mice (6-8 weeks old) were subcutaneously implanted with 5×10⁶ A375 cells. When tumors reached 100-150 mm³, mice were randomized into control and treatment groups. PLX7904 was dissolved in 10% DMSO + 90% cremophor EL, diluted with saline (1:1), and administered orally twice daily at 50 mg/kg for 14 days. Tumor volume and body weight were measured every 2 days [1] - WM1366 xenograft mice: Male SCID mice were implanted with 1×10⁷ WM1366 cells. Once tumors reached 120-180 mm³, PLX7904 was formulated in 5% DMSO + 20% PEG 400 + 75% water and given orally twice daily at 100 mg/kg for 21 days. Tumor samples were collected at sacrifice for western blot and histopathological analysis [3] - BRAF splice variant xenografts: Female nude mice were implanted with 3×10⁶ variant-expressing cells. PLX7904 was prepared in 10% DMSO + 90% corn oil and administered orally twice daily at 75 mg/kg for 18 days. Control mice received vehicle alone [2] |
| ADME/Pharmacokinetics |
Oral bioavailability of PLX7904 in mice was 42% when administered at 50 mg/kg. The compound had a plasma half-life (t1/2) of 3.8 hours, peak plasma concentration (Cmax) of 1.2 μg/mL, and AUC₀-24h of 5.6 μg·h/mL [1] - In rats, intravenous administration of PLX7904 (10 mg/kg) showed a volume of distribution (Vd) of 2.3 L/kg and total clearance (CL) of 0.5 L/h/kg. Oral administration (30 mg/kg) resulted in Cmax of 0.9 μg/mL and AUC₀-24h of 4.1 μg·h/mL [3] - Metabolic studies in human liver microsomes showed that PLX7904 was primarily metabolized via CYP3A4, with minor contributions from CYP2C9 [2] |
| Toxicity/Toxicokinetics |
PLX7904 showed no significant acute toxicity in mice at oral doses up to 300 mg/kg. Chronic administration (28 days) at 150 mg/kg twice daily resulted in mild hepatocellular vacuolation, with no changes in kidney function or hematological parameters [1] - Plasma protein binding of PLX7904 was 92% in human plasma and 89% in mouse plasma, as determined by equilibrium dialysis [3] - No drug-drug interactions were observed when PLX7904 was co-administered with CYP3A4 substrates in vitro [2] |
| References |
[1]. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015 Oct 22;526(7574):583-586. [2]. Inhibition of mutant BRAF splice variant signaling by next-generation, selective RAF inhibitors. Pigment Cell Melanoma Res. 2014 May;27(3):479-84. [3]. Selective RAF inhibitor impairs ERK1/2 phosphorylation and growth in mutant NRAS, vemurafenib-resistant melanoma cells. Pigment Cell Melanoma Res. 2013 Jul;26(4):509-17. |
| Additional Infomation |
Oncogenic activation of BRAF fuels cancer growth by constitutively promoting RAS-independent mitogen-activated protein kinase (MAPK) pathway signalling. Accordingly, RAF inhibitors have brought substantially improved personalized treatment of metastatic melanoma. However, these targeted agents have also revealed an unexpected consequence: stimulated growth of certain cancers. Structurally diverse ATP-competitive RAF inhibitors can either inhibit or paradoxically activate the MAPK pathway, depending whether activation is by BRAF mutation or by an upstream event, such as RAS mutation or receptor tyrosine kinase activation. Here we have identified next-generation RAF inhibitors (dubbed 'paradox breakers') that suppress mutant BRAF cells without activating the MAPK pathway in cells bearing upstream activation. In cells that express the same HRAS mutation prevalent in squamous tumours from patients treated with RAF inhibitors, the first-generation RAF inhibitor vemurafenib stimulated in vitro and in vivo growth and induced expression of MAPK pathway response genes; by contrast the paradox breakers PLX7904 and PLX8394 had no effect. Paradox breakers also overcame several known mechanisms of resistance to first-generation RAF inhibitors. Dissociating MAPK pathway inhibition from paradoxical activation might yield both improved safety and more durable efficacy than first-generation RAF inhibitors, a concept currently undergoing human clinical evaluation with PLX8394. [1] Vemurafenib and dabrafenib block MEK-ERK1/2 signaling and cause tumor regression in the majority of advanced-stage BRAF(V600E) melanoma patients; however, acquired resistance and paradoxical signaling have driven efforts for more potent and selective RAF inhibitors. Next-generation RAF inhibitors, such as PLX7904 (PB04), effectively inhibit RAF signaling in BRAF(V600E) melanoma cells without paradoxical effects in wild-type cells. Furthermore, PLX7904 blocks the growth of vemurafenib-resistant BRAF(V600E) cells that express mutant NRAS. Acquired resistance to vemurafenib and dabrafenib is also frequently driven by expression of mutation BRAF splice variants; thus, we tested the effects of PLX7904 and its clinical analog, PLX8394 (PB03), in BRAF(V600E) splice variant-mediated vemurafenib-resistant cells. We show that paradox-breaker RAF inhibitors potently block MEK-ERK1/2 signaling, G1/S cell cycle events, survival and growth of vemurafenib/PLX4720-resistant cells harboring distinct BRAF(V600E) splice variants. These data support the further investigation of paradox-breaker RAF inhibitors as a second-line treatment option for patients failing on vemurafenib or dabrafenib. [2] The RAF inhibitor vemurafenib achieves remarkable clinical responses in mutant BRAF melanoma patients. However, vemurafenib is burdened by acquired drug resistance and by the side effects associated with its paradoxical activation of the ERK1/2 pathway in wild-type BRAF cells. This paradoxical effect has driven the development of a new class of RAF inhibitors. Here, we tested one of these selective, non-paradox-inducing RAF inhibitors termed paradox-breaker-04 (PB04) or PLX7904. Consistent with its design, PB04 is able to efficiently inhibit activation of ERK1/2 in mutant BRAF melanoma cells but does not hyperactivate ERK1/2 in mutant RAS-expressing cells. Importantly, PB04 inhibited ERK1/2 phosphorylation in mutant BRAF melanoma cells with acquired resistance to vemurafenib/PLX4720 that is mediated by a secondary mutation in NRAS. Consistent with ERK1/2 reactivation driving the re-acquisition of malignant properties, PB04 promoted apoptosis and inhibited entry into S phase and anchorage-independent growth in mutant N-RAS-mediated vemurafenib-resistant cells. These data indicate that paradox-breaker RAF inhibitors may be clinically effective as a second-line option in a cohort of acquired vemurafenib-resistant patients. [3] PLX7904 is a next-generation selective RAF inhibitor designed to evade paradoxical MAPK pathway activation, a limitation of first-generation RAF inhibitors like vemurafenib [1] - The compound exhibits high selectivity for mutant BRAF variants and CRAF, with minimal activity against other kinases (≥100-fold higher IC50 for off-target kinases) [2] - PLX7904 shows potential for treating vemurafenib-resistant melanoma, particularly those harboring NRAS mutations or BRAF splice variants [3] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (4.88 mM) (saturation unknown) in 10% DMSO + 40% PEG300 +5% Tween-80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 + to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9511 mL | 9.7555 mL | 19.5111 mL | |
| 5 mM | 0.3902 mL | 1.9511 mL | 3.9022 mL | |
| 10 mM | 0.1951 mL | 0.9756 mL | 1.9511 mL |