PHA-793887 (PHA793887; PHA 793887) is a novel and ATP-competitive inhibitor of the multi-CDK (cyclin dependent kinases) for CDK2, CDK5 and CDK7 with potential anticancer activity. With IC50 values of 8 nM, 5 nM, and 10 nM, it inhibits CDK2/5/7.
Physicochemical Properties
| Molecular Formula | C19H31N5O2 | |
| Molecular Weight | 361.48 | |
| Exact Mass | 361.247 | |
| Elemental Analysis | C, 63.13; H, 8.64; N, 19.37; O, 8.85 | |
| CAS # | 718630-59-2 | |
| Related CAS # | 718630-60-5 (HCl);718630-59-2; | |
| PubChem CID | 46191454 | |
| Appearance | White Solid powder | |
| Density | 1.2±0.1 g/cm3 | |
| Boiling Point | 596.2±50.0 °C at 760 mmHg | |
| Flash Point | 314.4±30.1 °C | |
| Vapour Pressure | 0.0±1.7 mmHg at 25°C | |
| Index of Refraction | 1.573 | |
| LogP | 1.79 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 4 | |
| Rotatable Bond Count | 4 | |
| Heavy Atom Count | 26 | |
| Complexity | 542 | |
| Defined Atom Stereocenter Count | 0 | |
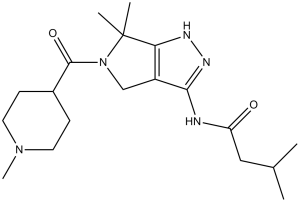
| SMILES | O=C(C1([H])C([H])([H])C([H])([H])N(C([H])([H])[H])C([H])([H])C1([H])[H])N1C([H])([H])C2C(N([H])C(C([H])([H])C([H])(C([H])([H])[H])C([H])([H])[H])=O)=NN([H])C=2C1(C([H])([H])[H])C([H])([H])[H] |
|
| InChi Key | HUXYBQXJVXOMKX-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C19H31N5O2/c1-12(2)10-15(25)20-17-14-11-24(19(3,4)16(14)21-22-17)18(26)13-6-8-23(5)9-7-13/h12-13H,6-11H2,1-5H3,(H2,20,21,22,25) | |
| Chemical Name | N-[6,6-dimethyl-5-(1-methylpiperidine-4-carbonyl)-1,4-dihydropyrrolo[3,4-c]pyrazol-3-yl]-3-methylbutanamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Cdk5/p25 (IC50 = 5 nM); cdk2/cyclin A (IC50 = 8 nM); CDK2/cyclinE (IC50 = 8 nM); CDK7/cyclin H (IC50 = 10 nM); Cdk1/cyclin B (IC50 = 60 nM); Cdk4/cyclin D1 (IC50 = 62 nM); CDK9/cyclinT1 (IC50 = 138 nM); GSK-3β (IC50 = 79 nM) | |
| ln Vitro |
|
|
| ln Vivo |
|
|
| Enzyme Assay | After a compound is incubated with particular enzymes and substrates, the phosphorylated product is quantified to ascertain the biochemical activity of the compound. The PHA-793887 (1.5 nM–10 μM) is incubated for 30−90 min at room temperature with a final volume of 30 μL of kinase buffer, substrate, and the specific enzyme (0.7−100 nM). The plates used are 96 U bottom plates. The reaction is halted after incubation, and the phosphorylated substrate is separated from nonincorporated radioactive ATP using Dowex resin, SPA beads, or Multiscreen phosphocellulose filters in the manner described below: (1) Regarding SPA Assays. One milligram of streptavidin-coated SPA beads is added to 100 microliters of PBS + 32 milligrams of EDTA + 0.1% Triton X-100 + 500 micrograms of ATP to halt the reaction. Following a twenty-minute incubation period for substrate capture, 100 microliters of the reaction mixture are poured into 96-well Optiplate plates that contain 100 microliters of 5 M CsCl. The plates are then allowed to stand for four hours to enable the beads to stratify to the top of the plate, and the amount of substrate-incorporated phosphate is measured using TopCount. (2) Regarding the Dowex Resin Assay. To halt the reaction and extract the unreacted 33P-γ-ATP and separate it from the phosphorylated substrate in solution, 150 μL of resin/formate, pH 3.00, is added. 50 μL of supernatant is transferred to Optiplate 96-well plates after a 60-minute rest period. Following the addition of 150 μL of Microscint 40, the radioactivity is measured using TopCount.(3) Regarding Multiscreen Assay. Addition of 10 μL of 150 mM EDTA stops the reaction. To enable substrate binding to the phosphocellulose filter, 100 μL is added to a MultiScreen plate. After that, the plates are dried and three times cleaned with 100 μL of H2PO4 (75 mM). Following the addition of 100 μL of Microscint 0, radioactivity is measured using TopCount. Through nonlinear regression analysis, IC50 values are found. | |
| Cell Assay | To conduct cytotoxicity assays, Alamar blue vital dye is used. Preliminary dose-response curves are carried out for every cell line to determine the cell-concentration range and provide a linear relationship with fluorescence. For cell lines, 200 μL of complete medium is plated in 96-well plates with 5,000–20,000 cells, either with or without increasing drug dosages (0.01–10 μM). 10 × 105 cells/well are plated in StemSpanSFEM medium and treated with the same range of drug concentrations for ALL-2 and AML-PS leukemias. Plates of 1 × 105 cells/well are made using cord blood CD34+ cells and peripheral blood mononuclear cells. The cells are treated with or without 1 μg/mL phytohemagglutin or a growth factor cocktail, which includes 50 ng/mL stem cell factor, 20 ng/mL granulocyte-macrophage colony-stimulating factor, granulocyte colony-stimulating factor, interleukin-3, interleukin-6, and 3 U/mL erythropoietin. All cases involve adding 1/10 volume Alamar blue solution and incubating overnight following a 48-hour culture. Then, using an excitation at 535 nm and an emission at 590 nm, the plates are read in a fluorimeter. When background fluorescence in the absence of cells is subtracted, the percentage of fluorescence relative to the untreated control is used to calculate cytotoxicity. | |
| Animal Protocol | SCID mice receive a subcutaneous inoculation with 107 HL60 and K562 cells. Seven mice per group are randomly assigned to the animals. In the HL60 model, PHA-793887 is given intravenously (IV) once daily at a dose of 20 mg/kg for ten days, from day 9 to day 18, and in two 5-day cycles (day 9 to day 13 and day 17 to day 21) in K562-bearing mice. Starting on day 9, Glivec is given orally for nine days in a row in the K562 xenograft model. Twice a week, net body weight and tumor growth are assessed. Tumor weight = length (mm) × width2 (mm) /2 is the formula used to calculate the weight of the tumor. The anticancer treatment's impact is measured by how long it takes for tumors to begin growing exponentially. The difference between the median time (in days) needed for the tumors in the treatment group (T) and the control group (C) to reach a predetermined size is known as the delay (T − C value). The reduction in body weight is the basis for evaluating toxicity. | |
| References |
[1]. Transcriptional analysis of an E2F gene signature as a biomarker of activity of the cyclin-dependent kinase inhibitor PHA-793887 in tumor and skin biopsies from a phase I clinical study. Mol Cancer Ther. 2010 May;9(5):1265-73. [2]. A first in man, phase I dose-escalation study of PHA-793887, an inhibitor of multiple cyclin-dependent kinases (CDK2, 1 and 4) reveals unexpected hepatotoxicity in patients with solid tumors. Cell Cycle. 2011 Mar 15;10(6):963-70. Epub 2011 Mar 15. [3]. Therapeutic efficacy of the pan-cdk inhibitor PHA-793887 in vitro and in vivo in engraftment and high-burden leukemia models. Exp Hematol. 2010 Apr;38(4):259-269.e2. [4]. Optimization of 6,6-dimethyl pyrrolo[3,4-c]pyrazoles: Identification of PHA-793887, a potent CDK inhibitor suitable for intravenous dosing. Bioorg Med Chem. 2010 Mar 1;18(5):1844-53. |
|
| Additional Infomation |
N-[6,6-dimethyl-5-[(1-methyl-4-piperidinyl)-oxomethyl]-1,4-dihydropyrrolo[3,4-c]pyrazol-3-yl]-3-methylbutanamide is a piperidinecarboxamide. PHA-793887 has been used in trials studying the treatment of Advanced/Metastatic Solid Tumors. |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (6.92 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 2: 30% Propylene glycol , 5% Tween 80 , 65% D5W: 15 mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7664 mL | 13.8320 mL | 27.6640 mL | |
| 5 mM | 0.5533 mL | 2.7664 mL | 5.5328 mL | |
| 10 mM | 0.2766 mL | 1.3832 mL | 2.7664 mL |