PHA-767491 HCl (formerly known as CAY10572 HCl) is a novel potent ATP-competitive and dual CDC7/CDK9 inhibitor with IC50 of 10 nM and 34 nM in cell-free assays, respectively. It exhibits selectivity of approximately 20 times against CDK1/2 and GSK3-β, 50 times against MK2 and CDK5, and 100 times against PLK1 and CHK2. An important kinase called CDC7 stimulates replication origins to facilitate DNA replication. PHA-767491 inhibits the synthesis of DNA and modifies the replicative DNA helicase's phosphorylation at CDC7-dependent phosphorylation sites. PHA-767491, in contrast to existing DNA synthesis inhibitors, inhibits replication origin activation without impeding replication fork progression or causing a prolonged DNA damage response. In preclinical cancer models, PHA-767491 treatment induces apoptotic cell death in a variety of cancer cell types and inhibits tumor growth. PHA-767491 is the first known molecule to directly influence the mechanisms governing initiation rather than elongation in DNA replication, and its actions imply that inhibiting Cdc7 kinase may be a novel approach to the development of anticancer treatments.
Physicochemical Properties
| Molecular Formula | C12H11N3O.HCL | |
| Molecular Weight | 249.7 | |
| Exact Mass | 249.067 | |
| Elemental Analysis | C, 57.72; H, 4.84; Cl, 14.20; N, 16.83; O, 6.41. | |
| CAS # | 942425-68-5 | |
| Related CAS # | PHA-767491;845714-00-3; PHA-767491 hydrochloride;942425-68-5; 845538-12-7 (2HCl) | |
| PubChem CID | 11715766 | |
| Appearance | Light yellow to yellow solid powder | |
| LogP | 2.493 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 2 | |
| Rotatable Bond Count | 1 | |
| Heavy Atom Count | 17 | |
| Complexity | 275 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | Cl.O=C1C2=C(NC(C3C=CN=CC=3)=C2)CCN1 |
|
| InChi Key | IMVNFURYBZMFDZ-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C12H11N3O.ClH/c16-12-9-7-11(8-1-4-13-5-2-8)15-10(9)3-6-14-12;/h1-2,4-5,7,15H,3,6H2,(H,14,16);1H | |
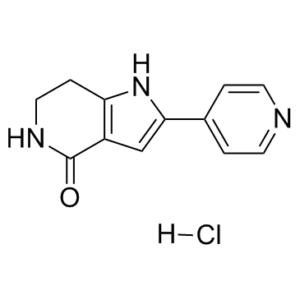
| Chemical Name | 2-pyridin-4-yl-1,5,6,7-tetrahydropyrrolo[3,2-c]pyridin-4-one;hydrochloride | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | CDK9 (IC50 = 34 nM); CDK2 (IC50 = 240 nM); CDK1 (IC50 = 250 nM); CDK5 (IC50 = 460 nM); GSK3-β (IC50 = 220 nM); Mk2 (IC50 = 470 nM); Plk1 (IC50 = 980 nM); Chk2 (IC50 = 1100 nM) | ||
| ln Vitro | PHA-767491 exhibits selectivity of about 20-fold for Cdk1, Cdk2, and GSK3-β, 50-fold for MK2 and Cdk5, and 100-fold for PLK1 and CHK2. Unlike 5-FU or gemcitabine, which only works in a few cell lines, PHA-767491 significantly induces apoptosis in a p53-independent manner in almost all cell lines. It also inhibits cell proliferation in a variety of human cell lines, with IC50 values ranging from 0.86 μM for SF-268 to 5.87 μM for K562. PHA-767491 treatment at 5 μM specifically inhibits Cdc7 kinase and Mcm2 phosphorylation at the Cdc7-dependent Ser40 site, blocking the initiation of DNA replication but not replication fork progression, in contrast to current DNA synthesis inhibitors.[1] PHA-767491 treatment at 3 μM can significantly reduce the up-regulated Mcl-1 levels in ABT-737-resistant OCI-LY1 and SU-DHL-4 cells, possibly through Cdk9 inhibition, restoring the sensitivity to ABT-737. **[2]** When PHA-767491 is applied at 1 μM, it also causes direct mitochondrial dependent pro-apoptosis in quiescent chronic lymphocytic leukemia (CLL) cells via a similar mechanism (EC50 of 0.34-0.97 μM). PHA-767491 treatment at 5 μM inhibits Cdc7 instead of causing cell death in proliferating CLL cells stimulated by CD154 and interleukin-4, which results in the elimination of DNA synthesis. | ||
| ln Vivo |
PHA-767491 administered twice daily for five days markedly inhibits the growth of HL60 xenograft in a dose-dependent manner with TGI of 50% and 92% at doses of 20 mg/kg and 30 mg/kg, respectively. This effect is also evident in A2780, Mx-1, and HCT-116 xenograft models and the DMBA-induced mammary carcinomas. It is associated with decreased phosphorylation of Mcm2 at the Cdc7-dependent site Ser40 [1]. PHA-767491 promotes in situ cell apoptosis and reduces Chk1 phosphorylation in tumor tissues sectioned from naked mice HCC xenografts[2]. PHA-767491 has antitumor activity in cancer models [4] The potential of PHA-767491 as an anticancer drug was first evaluated in nude mice carrying subcutaneous implanted tumors derived from the acute myeloid leukaemia (AML) HL60 human cell line. After intravenous administration at two dose levels of 20 and 30 mg kg−1 twice a day, for five consecutive days, a dose-dependent reduction in tumor volume with respect to vehicle-treated animals was observed (Fig. 4a). Tumor growth inhibition, calculated the day after the end of treatment, was 50% at the lower dose, and 92% at the higher dose, where evidence of tumor regression in five out of eight animals was observed. Under these conditions the compound reached micromolar plasma levels, which is consistent with active levels in cell-based assays, with an area under the concentration-time curve (AUC) of 47 μM h−1 and 71 μM h−1, respectively. PHA-767491 showed a good volume of distribution in tissues (approximately twice the total body water content) and was rapidly cleared from plasma (Supplementary Fig. 7 online). At these doses the compound appeared to be well tolerated, and it did not cause significant body weight loss; however, a further dose escalation was not tolerated. In a toxicology study in which PHA-767491 was administered for 5 d at 30 mg kg−1 twice a day, no clinical signs or gross lesions were observed. Histopathological analysis of 36 different organs explanted from the treated animals indicated signs of atrophy of the testes, moderate myeloid hyperplasia in the bone marrow and minimal lymphoid depletion in the spleen, which is consistent with the reported high levels of Cdc7 expression in testis10 and with Cdc7's role in highly proliferating tissues. The administration of PHA-767491 also resulted in tumor growth inhibition in the A2780 ovary carcinoma, in Mx-1 mammary adenocarcinoma and in HCT-116 colon carcinoma xenograft models, with a tumor growth inhibition of approximately 50% measured after the 5 d of treatment (Fig. 4b and Supplementary Fig. 8 online). We then administered PHA-767491 to rats with 7,12-dimethylbenz(a)anthracene (DMBA, 12)-induced mammary carcinomas for 10 d. In this experiment tumor growth was suppressed during the treatment and strongly reduced for a further two weeks (Fig. 4c). In order to correlate the antitumor activity with Cdc7 inhibition, HCT-116 tumors explanted from controls or animals treated with a 5-d cycle of PHA-767491 were analyzed by western blot. Phosphorylation of Mcm2 at the Cdc7-dependent site Ser40 was greatly decreased in the tumors of treated animals (Fig. 5a). Immunohistochemistry (IHC) of tumor sections confirmed lower levels of Ser40 Mcm2 phosphorylation in most of the cells of the treated tumor's viable areas (Fig. 5b), whereas the levels of Rb phosphorylation at Ser807/811 and the numbers of cyclin A–positive cells were not decreased. PHA-767491 treatment caused a marked increase of Ki67-positive cells for reasons not yet understood. Altogether these results indicate that (i) PHA-767491 can inhibit Cdc7 kinase in vivo and that (ii) the loss of Mcm2 phosphorylation is a direct effect of the compound on viable cycling cells, and is not caused by a decreased proliferation index in treated tumor cells, or by the differential presence of areas of necrosis—a characteristic of HCT-116–derived xenograft tumors38. We conclude that PHA-767491 has antitumor activity in vivo in multiple preclinical cancer models and in at least two different species. |
||
| Enzyme Assay |
Increasing concentrations of each DDK inhibitor are pre-incubated for five minutes with 20 ng of purified human DDK. After adding 1.5 µM cold ATP and 10 µCi (γ)-32P ATP, the mixture is mixed with 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, and 1 mM DTT. It is then incubated for 30 minutes at 30°C. SDS-PAGE and autoradiography on HyBlot CL film are performed after the proteins are denatured in 1X Laemmli buffer at 100°C. One way to measure the kinase activity of DDK is to look for auto-phosphorylation. ImageJ is used to quantify 32P-labeled bands, and GraphPad is used to determine the IC50 values. In vitro kinase assays.[4] The potency of the compound toward Cdc7 and 37 additional kinases belonging to our kinase selectivity screening (KSS) panel was determined using either a strong anion exchanger (Dowex 1-X8 resin, formate form)-based assay or a scintillation proximity assay, as previously described25,26. Cdk9 activity was measured using 50 nM of recombinant Cdk9/cyclin T in 50 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM DTT, 3 μM Na3VO4, 150 μM RNA polymerase CDT peptide and 80 μM ATP. Cdk7 assay was performed in the same buffer using 37 nM of purified kinase in the presence of 200 μM ATP and 10 μM myelin binding protein as a substrate. For each enzyme, the absolute Km values for ATP and the specific substrate were initially determined, and each assay was then run at optimized ATP (2Km) and substrate (5Km) concentrations. Because under these conditions IC50 = 3βKi, this setting enabled direct comparison of IC50 values of PHA-767491 across the KSS panel for the evaluation of its biochemical selectivity. |
||
| Cell Assay |
There are 2500 cells plated in each well of 96-well plates used for assays. Cells undergo treatment with small molecule inhibitors after 24 hours, and they are then incubated at 37°C for 72 hours. Next, the cells undergo lysis, and the CellTiter-Glo assay is employed to quantify the ATP content, which serves as a marker of metabolically active cells. Utilizing GraphPad software, IC50 values are determined. 100,000 cells are plated per well in six-well plates used for assays. Small molecule inhibitors are applied to the cells after a day, and they are then cultured for different lengths of time. Trypsinized cells are suspended in 5 milliliters of phosphate-buffered saline. After mixing 30 µL of this suspension with 30 µL of CellTiter-Glo reagent, it is incubated at room temperature for 10 minutes. The EnVision 2104 Multilabel Reader and the BioTek Synergy Neo Microplate Reader are used to measure luminosity.
Cell viability assay [3] 5×103 U87-MG and U251-MG cells were seeded in a 96-well plate 24 h before treatment. Next day, cells were treated with inhibitor (10 µM final concentration), solvent control (water), or left untreated. Seventy-two hours after treatment, 10 µl of PrestoBlue cell viability reagent was added onto the cells to assess cell viability. Relative cell viability was calculated by setting the viability of solvent control as 100%. Experiments were repeated at least three times. Cell proliferation assay [3] For synchronization, U87-MG and U251-MG cells were maintained in culture medium supplemented with 1% FBS for 24 h. Then, 1 × 104 U87-MG and U251-MG cells were seeded in a 96-well plate. Next day, cells were treated with inhibitor (2.5 or 10 µM final concentration), solvent control (water), or left untreated. Seventy-two hours after treatment, bromodeoxyuridine (BrdU) cell proliferation ELISA kit was used according to the manufacturer’s instructions. Rate of proliferation in cells treated with solvent control was set as 100% to calculate relative cell proliferation rate. |
||
| Animal Protocol |
|
||
| Toxicity/Toxicokinetics | In a toxicology study in which PHA-767491 was administered for 5 d at 30 mg kg−1 twice a day, no clinical signs or gross lesions were observed. Histopathological analysis of 36 different organs explanted from the treated animals indicated signs of atrophy of the testes, moderate myeloid hyperplasia in the bone marrow and minimal lymphoid depletion in the spleen, which is consistent with the reported high levels of Cdc7 expression in testis10 and with Cdc7's role in highly proliferating tissues. [4] | ||
| References |
[1]. The potent Cdc7-Dbf4 (DDK) kinase inhibitor XL413 has limited activity in many cancer cell lines and discovery of potential new DDK inhibitor scaffolds. PLoS One. 2014 Nov 20;9(11):e113300. [2]. Dual Inhibition of Cdc7 and Cdk9 by PHA-767491 Suppresses Hepatocarcinoma Synergistically with 5-Fluorouracil. Curr Cancer Drug Targets. 2015;15(3):196-204. [3]. Cell division cycle 7-kinase inhibitor PHA-767491 hydrochloride suppresses glioblastoma growth and invasiveness. Cancer Cell Int. 2016 Nov 18;16:88. [4]. A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat Chem Biol. 2008 Jun;4(6):357-65. |
||
| Additional Infomation |
2-pyridin-4-yl-1,5,6,7-tetrahydropyrrolo[3,2-c]pyridin-4-one is a pyrrolopyridine. PHA-767491 is a Cdc7/CDK9 inhibitor. Cdc7-Dbf4 kinase or DDK (Dbf4-dependent kinase) is required to initiate DNA replication by phosphorylating and activating the replicative Mcm2-7 DNA helicase. DDK is overexpressed in many tumor cells and is an emerging chemotherapeutic target since DDK inhibition causes apoptosis of diverse cancer cell types but not of normal cells. PHA-767491 and XL413 are among a number of potent DDK inhibitors with low nanomolar IC50 values against the purified kinase. Although XL413 is highly selective for DDK, its activity has not been extensively characterized on cell lines. We measured anti-proliferative and apoptotic effects of XL413 on a panel of tumor cell lines compared to PHA-767491, whose activity is well characterized. Both compounds were effective biochemical DDK inhibitors but surprisingly, their activities in cell lines were highly divergent. Unlike PHA-767491, XL413 had significant anti-proliferative activity against only one of the ten cell lines tested. Since XL413 did not effectively inhibit DDK in multiple cell lines, this compound likely has limited bioavailability. To identify potential leads for additional DDK inhibitors, we also tested the cross-reactivity of ∼400 known kinase inhibitors against DDK using a DDK thermal stability shift assay (TSA). We identified 11 compounds that significantly stabilized DDK. Several inhibited DDK with comparable potency to PHA-767491, including Chk1 and PKR kinase inhibitors, but had divergent chemical scaffolds from known DDK inhibitors. Taken together, these data show that several well-known kinase inhibitors cross-react with DDK and also highlight the opportunity to design additional specific, biologically active DDK inhibitors for use as chemotherapeutic agents.[1] Activation of checkpoint kinase 1 (Chk1) is essential in chemoresistance of hepatocarcinoma (HCC) to 5-fluorouracil (5-FU) and other antimetabolite family of drugs. In this study, we demonstrated that PHA-767491, a dual inhibitor of two cell cycle checkpoint kinases, cell division cycle kinase 7 (Cdc7) and cyclin-dependent kinase 9 (Cdk9), has synergistic antitumor effect with 5-FU to suppress human HCC cells both in vitro and in vivo. Compared with the sole use of each agent, PHA-767491 in combination with 5-FU exhibited much stronger cytotoxicity and induced significant apoptosis manifested by remarkably increased caspase 3 activation and poly(ADP-Ribose) polymerase fragmentation in HCC cells. PHA-767491 directly counteracted the 5-FU-induced phosphorylation of Chk1, a substrate of Cdc7; and decreased the expression of the anti-apoptotic protein myeloid leukemia cell 1, a downstream target of Cdk9. In tumor tissues sectioned from nude mice HCC xenografts, administration of PHA-767491 also decreased Chk1 phosphorylation and increased in situ cell apoptosis. Our study suggests that PHA- 767491 could enhance the efficacy of 5-FU by inhibiting Chk1 phosphorylation and down-regulating Mcl1 expression through inhibition of Cdc7 and Cdk9, thus combinational administration of PHA-767491 with 5-FU could be potentially beneficial to patients with advanced and resistant HCC. [2] Background: Genomic instability is a hallmark of cancer cells, and this cellular phenomenon can emerge as a result of replicative stress. It is possible to take advantage of replicative stress, and enhance it in a targeted way to fight cancer cells. One of such strategies involves targeting the cell division cycle 7-related protein kinase (CDC7), a protein with key roles in regulation of initiation of DNA replication. CDC7 overexpression is present in different cancers, and small molecule inhibitors of the CDC7 have well-documented anti-tumor effects. Here, we aimed to test the potential of CDC7 inhibition as a new strategy for glioblastoma treatment. Methods: PHA-767491 hydrochloride was used as the CDC7 inhibitor. Two glioblastoma cell lines (U87-MG and U251-MG) and a control cell line (3T3) were used to characterize the effects of CDC7 inhibition. The effect of CDC7 inhibition on cell viability, cell proliferation, apoptosis, migration, and invasion were analyzed. In addition, real-time PCR arrays were used to identify the differentially expressed genes in response to CDC7 inhibition. Results: Our results showed that CDC7 inhibition reduces glioblastoma cell viability, suppresses cell proliferation, and triggers apoptosis in glioblastoma cell lines. In addition, we determined that CDC7 inhibition also suppresses glioblastoma cell migration and invasion. To identify molecular targets of CDC7 inhibition, we used real-time PCR arrays, which showed dysregulation of several mRNAs and miRNAs. Conclusions: Taken together, our findings suggest that CDC7 inhibition is a promising strategy for treatment of glioblastoma.[3] Cdc7 is an essential kinase that promotes DNA replication by activating origins of replication. Here, we characterized the potent Cdc7 inhibitor PHA-767491 (1) in biochemical and cell-based assays, and we tested its antitumor activity in rodents. We found that the compound blocks DNA synthesis and affects the phosphorylation of the replicative DNA helicase at Cdc7-dependent phosphorylation sites. Unlike current DNA synthesis inhibitors, PHA-767491 prevents the activation of replication origins but does not impede replication fork progression, and it does not trigger a sustained DNA damage response. Treatment with PHA-767491 results in apoptotic cell death in multiple cancer cell types and tumor growth inhibition in preclinical cancer models. To our knowledge, PHA-767491 is the first molecule that directly affects the mechanisms controlling initiation as opposed to elongation in DNA replication, and its activities suggest that Cdc7 kinase inhibition could be a new strategy for the development of anticancer therapeutics. [4] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 1 mg/mL (4.00 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 10.0 mg/mL clear DMSO stock solution to 400 μL of PEG300 and mix evenly; then add 50 μL of Tween-80 to the above solution and mix evenly; then add 450 μL of normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 1 mg/mL (4.00 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 10.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: 5% DMSO+30% PEG 300+2% Tween 80+ddH2O: 1mg/mL Solubility in Formulation 4: 50 mg/mL (200.24 mM) in PBS (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.0048 mL | 20.0240 mL | 40.0481 mL | |
| 5 mM | 0.8010 mL | 4.0048 mL | 8.0096 mL | |
| 10 mM | 0.4005 mL | 2.0024 mL | 4.0048 mL |