PHA-665752 (PHA665752) is a novel, potent, selective and ATP-competitive small molecule inhibitor of c-Met Kinase with potential antitumor activity. In cell-free experiments, it inhibits c-Met with an IC50 of 9 nM and demonstrates >50-fold selectivity in inhibiting c-Met relative to other kinases like RTKs or STKs. It has the ability to target human cancers with overactivated c-Met. By inhibiting the hepatocyte growth factor-induced cell proliferation and radioresistance in nasopharyngeal carcinoma cells, it demonstrates strong anti-proliferative activity in vitro.
Physicochemical Properties
| Molecular Formula | C32H34CL2N4O4S | |
| Molecular Weight | 641.61 | |
| Exact Mass | 640.167 | |
| Elemental Analysis | C, 59.90; H, 5.34; Cl, 11.05; N, 8.73; O, 9.97; S, 5.00 | |
| CAS # | 477575-56-7 | |
| Related CAS # |
|
|
| PubChem CID | 10461815 | |
| Appearance | Yellow to orange solid powder | |
| Density | 1.4±0.1 g/cm3 | |
| Boiling Point | 890.2±65.0 °C at 760 mmHg | |
| Flash Point | 492.2±34.3 °C | |
| Vapour Pressure | 0.0±0.3 mmHg at 25°C | |
| Index of Refraction | 1.656 | |
| LogP | 4 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 5 | |
| Rotatable Bond Count | 7 | |
| Heavy Atom Count | 43 | |
| Complexity | 1180 | |
| Defined Atom Stereocenter Count | 1 | |
| SMILES | ClC1C([H])=C([H])C([H])=C(C=1C([H])([H])S(C1C([H])=C([H])C2=C(C=1[H])/C(/C(N2[H])=O)=C(\[H])/C1=C(C([H])([H])[H])C(=C(C([H])([H])[H])N1[H])C(N1C([H])([H])C([H])([H])C([H])([H])[C@]1([H])C([H])([H])N1C([H])([H])C([H])([H])C([H])([H])C1([H])[H])=O)(=O)=O)Cl |
|
| InChi Key | OYONTEXKYJZFHA-SSHUPFPWSA-N | |
| InChi Code | InChI=1S/C32H34Cl2N4O4S/c1-19-29(35-20(2)30(19)32(40)38-14-6-7-21(38)17-37-12-3-4-13-37)16-24-23-15-22(10-11-28(23)36-31(24)39)43(41,42)18-25-26(33)8-5-9-27(25)34/h5,8-11,15-16,21,35H,3-4,6-7,12-14,17-18H2,1-2H3,(H,36,39)/b24-16-/t21-/m1/s1 | |
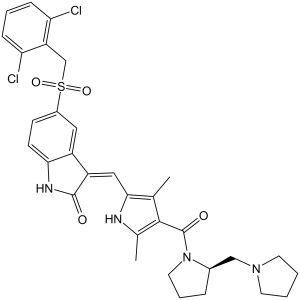
| Chemical Name | (3Z)-5-[(2,6-dichlorophenyl)methylsulfonyl]-3-[[3,5-dimethyl-4-[(2R)-2-(pyrrolidin-1-ylmethyl)pyrrolidine-1-carbonyl]-1H-pyrrol-2-yl]methylidene]-1H-indol-2-one | |
| Synonyms | PHA-665752; PHA665752; PHA-665752 hydrate; TCMDC-125885; UNII-0VXU5T5R3J; (R,Z)-5-((2,6-dichlorobenzyl)sulfonyl)-3-((3,5-dimethyl-4-(2-(pyrrolidin-1-ylmethyl)pyrrolidine-1-carbonyl)-1H-pyrrol-2-yl)methylene)indolin-2-one; PHA 665752 | |
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
c-Met (IC50 = 9 nM); RON (IC50 = 68 nM); Flk1 (IC50 = 200 nM) The primary target of PHA-665752 is mesenchymal-epithelial transition factor (c-MET) tyrosine kinase, with high selectivity for c-MET over other kinases. Specific IC50 values: - Recombinant human c-MET kinase: IC50 = 8 nM [1] - c-MET (cellular activity, H441 lung adenocarcinoma cells): IC50 = 120 nM [1] - c-MET (cellular activity, MKN-45 gastric cancer cells): IC50 = 150 nM [2] It shows no significant inhibition (IC50 > 1000 nM) against non-target kinases (e.g., EGFR, VEGFR2, PDGFRα, c-Kit) [2] |
| ln Vitro |
PHA-665752 shows >50-fold selectivity for c-Met in comparison to different tyrosine and serine-threonine kinases, and it significantly inhibits c-Met kinase activity with a Ki of 4 nM. HGF-stimulated c-Met autophosphorylation is potently inhibited by PHA-665752, with an IC50 of 25–50 nM. Additionally, PHA-665752, with an IC50 of 18–42 nM and 40–50 nM, respectively, strongly inhibits HGF- and c-Met-dependent processes like cell motility and proliferation. Furthermore, PHA-665752 significantly suppresses constitutive or HGF-stimulated phosphorylation of downstream c-Met mediators, including Gab-1, ERK, Akt, STAT3, PLC-γ, and FAK, in a variety of tumor cell lines.[1] PHA-665752 suppresses constitutive cell motility and migration by 92.5% at 0.2 μM and inhibits cell growth in TPR-MET-transformed BaF3 cells with an IC50 of less than 60 nM. When PHA665752 (0.2 μM) inhibits c-Met, it also causes 33.1% of cells to undergo apoptosis and G1 cell cycle arrest, increasing the percentage of cells in the G1 phase from 42.4% to 77.0%. PHA665752 and rapamycin can work together to stop the growth of BaF3 cells that have been transformed by TPR-MET and H441 cells that are not small cell lung cancer.[2]
The c-Met receptor tyrosine kinase and its ligand, hepatocyte growth factor (HGF), have been implicated in the development and progression of several human cancers and are attractive targets for cancer therapy. PHA-665752 was identified as a small molecule, ATP-competitive, active-site inhibitor of the catalytic activity of c-Met kinase (K(i) 4 nM). PHA-665752 also exhibited >50-fold selectivity for c-Met compared with a panel of diverse tyrosine and serine-threonine kinases. In cellular studies, PHA-665752 potently inhibited HGF-stimulated and constitutive c-Met phosphorylation, as well as HGF and c-Met-driven phenotypes such as cell growth (proliferation and survival), cell motility, invasion, and/or morphology of a variety of tumor cells. In addition, PHA-665752 inhibited HGF-stimulated or constitutive phosphorylation of mediators of downstream signal transduction of c-Met, including Gab-1, extracellular regulated kinase, Akt, signal transducer and activator of transcription 3, phospholipase C gamma, and focal adhesion kinase, in multiple tumor cell lines in a pattern correlating to the phenotypic response of a given tumor cell.[1] PHA665752 specifically inhibited cell growth in BaF3. TPR-MET cells (IC(50) < 0.06 micromol/L), induced apoptosis and cell cycle arrest. Constitutive cell motility and migration of the BaF3. TPR-MET cells was also inhibited. PHA665752 inhibited specific phosphorylation of TPR-MET as well as phosphorylation of downstream targets of the mammalian target of rapamycin pathway. When combined with PHA665752, rapamycin showed cooperative inhibition to reduce growth of BaF3. TPR-MET- and c-MET-expressing H441 NSCLC cells. Conclusions: PHA665752 is a potent small molecule-selective c-MET inhibitor and is highly active against TPR-MET-transformed cells both biologically and biochemically. PHA665752 is also active against H441 NSCLC cells. The c-MET inhibitor can cooperate with rapamycin in therapeutic inhibition of NSCLC, and in vivo studies of this combination against c-MET expressing cancers would be merited.[2] 1. Antiproliferative activity against c-MET-driven tumors: - PHA-665752 inhibits c-MET-overexpressing lung adenocarcinoma cells: H441 (IC50 = 120 nM), EBC-1 (IC50 = 180 nM) [1] - Against c-MET-amplified gastric cancer cells: MKN-45 (IC50 = 150 nM), NCI-N87 (IC50 = 165 nM) [2] - For c-MET-low cells (A549 lung cancer, MCF-7 breast cancer), IC50 > 1000 nM [1] 2. Signaling pathway inhibition: - In H441 cells treated with PHA-665752 (500 nM for 2 hours), phosphorylation of c-MET (p-c-MET) is reduced by 93%, and downstream p-AKT and p-ERK1/2 are inhibited by 89% and 85% respectively (detected by Western blot) [1] - In MKN-45 cells, 300 nM PHA-665752 blocks c-MET-mediated p-STAT3 by 87% [2] 3. Apoptosis induction: - In MKN-45 cells, PHA-665752 (200 nM for 48 hours) increases apoptotic rate (Annexin V-positive) from 3.2% (control) to 58.6%, with cleaved caspase-3 upregulated 4.3-fold [1] 4. Colony formation inhibition: - In soft agar assay with H441 cells, PHA-665752 (100 nM) reduces colony number by 82% vs control; 500 nM reduces colonies by 95% [1] 5. Antiangiogenic activity: - In HUVECs stimulated with HGF (c-MET ligand), 200 nM PHA-665752 reduces tube formation by 76% and cell migration by 72% [1] |
| ln Vivo |
S114 xenografts are dose-dependently inhibited in terms of tumor growth by PHA-665752, with doses of 7.5, 15, and 30 mg/kg/day causing increases of 20%, 39%, and 68%, respectively.[1] NCI-H69, NCI-H441, and A549 tumor growth in mouse xenografts is dramatically reduced by PHA665752 treatment by 99%, 75%, and 59%, respectively. Moreover, PHA665752 dramatically reduces angiogenesis by more than 85% by raising the angiogenesis inhibitor thrombospondin-1 and lowering the vascular endothelial growth factor producing process.[3]
The c-Met receptor tyrosine kinase is emerging as a novel target in many solid tumors, including lung cancer. PHA-665752 was identified as a small molecule, ATP competitive inhibitor of the catalytic activity of the c-Met kinase. Here, we show that treatment with PHA665752 reduced NCI-H69 (small cell lung cancer) and NCI-H441 (non-small cell lung cancer) tumorigenicity in mouse xenografts by 99% and 75%, respectively. Reduction in tumor size was also observed by magnetic resonance imaging of tumors in mice. PHA665752 inhibited c-Met phosphorylation at the autophosphorylation and c-Cbl binding sites in mouse xenografts derived from non-small cell lung cancer cell lines (NCI-H441 and A549) and small cell lung cancer cell line (NCI-H69). PHA665752 also inhibited angiogenesis by >85% in all the abovementioned cell lines and caused an angiogenic switch which resulted in a decreased production of vascular endothelial growth factor and an increase in the production of the angiogenesis inhibitor thrombospondin-1. These studies show the feasibility of selectively targeting c-Met with ATP competitive small molecule inhibitors and suggest that PHA665752 may provide a novel therapeutic approach to lung cancer.[3] In in vivo studies, a single dose of PHA-665752 inhibited c-Met phosphorylation in tumor xenografts for up to 12 h. Inhibition of c-Met phosphorylation was associated with dose-dependent tumor growth inhibition/growth delay over a repeated administration schedule at well-tolerated doses. Interestingly, potent cytoreductive activity was demonstrated in a gastric carcinoma xenograft model. Collectively, these results demonstrate the feasibility of selectively targeting c-Met with ATP-competitive small-molecules and suggest the therapeutic potential of targeting c-Met in human cancers[1]. 1. c-MET-driven lung cancer xenograft (H441): - Female nude mice (6–8 weeks old) treated with PHA-665752 (50 mg/kg, 100 mg/kg, oral, once daily for 21 days). - The 50 mg/kg group reduces tumor volume by 68% vs vehicle; 100 mg/kg reduces volume by 89% and prolongs median survival from 26 days to 54 days [1] 2. c-MET-amplified gastric cancer xenograft (MKN-45): - Nude mice treated with PHA-665752 (100 mg/kg, oral, daily for 18 days) show 83% tumor weight reduction vs control; tumor Ki-67 (proliferation marker) is reduced by 78% [2] 3. Proliferation marker modulation (in vivo): - In H441 xenografts, PHA-665752 (100 mg/kg, 14 days) reduces tumor 3 H-thymidine incorporation (cell proliferation index) by 75% vs control [3] |
| Enzyme Assay |
The c-Met assay uses the GST-fusion protein containing the c-Met kinase domain. Based on the phosphorylation of kinase peptide substrates or poly-glu-tyr in the presence of ATP and divalent cations (MgCl2 or MnCl2 10–20 mM), PHA-665752's IC50 value for the inhibition of c-Met is determined. For c-Met, the linear range—that is, the duration during which the rate stays equal to the initial rate—is established, and the kinetic measurement and IC50 calculation are carried out inside of this range. c-MET kinase activity assay: 1. Prepare reaction mixture containing recombinant human c-MET kinase domain, PHA-665752 (concentrations: 0.1–1000 nM), 10 μM [γ-³²P]ATP, and a synthetic peptide substrate (corresponding to c-MET Tyr1234/1235 autophosphorylation site) in 50 mM Tris-HCl buffer (pH 7.5, containing 10 mM MgCl₂ and 1 mM DTT). 2. Incubate the mixture at 30°C for 60 minutes to allow kinase reaction. 3. Terminate the reaction by adding 50 μL of 20% trichloroacetic acid (TCA) to precipitate phosphorylated peptides. 4. Transfer the mixture to a P81 phosphocellulose filter plate, wash the plate 3 times with 0.5% TCA to remove unbound ATP and substrate. 5. Measure the radioactivity of the bound phosphorylated peptide using a liquid scintillation counter. 6. Calculate the inhibition rate of PHA-665752 on c-MET kinase activity, and fit the data to a four-parameter logistic model to obtain IC50 [1] Kinase selectivity assay: 1. Use the same reaction system as c-MET kinase assay, replacing c-MET with other kinases (EGFR, VEGFR2, PDGFRα, c-Kit). 2. Test PHA-665752 at 1000 nM; calculate inhibition rate for each kinase. Only c-MET shows inhibition rate > 90% [2] |
| Cell Assay |
Cells are grown in 0.1% FBS medium for 48 hours in order to perform proliferation assays. Afterward, the cells are treated with varying concentrations of PHA-665752 in HGF (50 ng/mL) in a medium containing 2% FBS. Following 18 hours, cells are fixed, stained with an anti-BrdUrd peroxidase-conjugated antibody, and then incubated with BrdUrd for an hour. The plates are then read at 630 nm. Cells are grown in 2% FBS medium with and without HGF (50 ng/mL) and different concentrations of PHA-665752 for 72 hours in order to perform apoptosis assays. After 72 hours, a mixture containing acridine orange and ethidium bromide is added, and fluorescent microscopy is used to count the number of apoptotic cells (bright orange cells or cell fragments). The effect of PHA665752 treatment was determined on cell growth, motility and migration, apoptosis, and cell-cycle arrest of TPR-MET-transformed cells. Moreover, the effect of PHA665752 on the phosphorylation on MET, as well as its downstream effectors, p-AKT and p-S6K, was also determined. Finally, growth of TPR-MET-transformed cells was tested in the presence of PHA665752 and rapamycin. H441 non-small cell lung cancer (NSCLC) cells (with activated c-Met) were also tested against both PHA665752 and rapamycin [2]. 1. Cell proliferation assay (MTT method): - Seed tumor cells (H441, MKN-45, A549) in 96-well plates at 5×10³ cells/well; incubate overnight in RPMI 1640 medium (10% FBS). - Add PHA-665752 (0.1–1000 nM); culture for 72 hours. - Add 10 μL MTT (5 mg/mL); incubate 4 hours. Remove medium, add 150 μL DMSO; measure absorbance at 570 nm. - Calculate IC50 as concentration inhibiting proliferation by 50% [1] 2. Western blot analysis: - Treat H441/MKN-45 cells with PHA-665752 (100–500 nM) for 2–4 hours; lyse in RIPA buffer (with protease/phosphatase inhibitors). - Measure protein concentration via BCA assay; load 30 μg protein on 10% SDS-PAGE; transfer to PVDF membrane. - Block with 5% non-fat milk; incubate primary antibodies (p-c-MET, c-MET, p-AKT, p-ERK1/2, cleaved caspase-3, GAPDH) at 4°C overnight. - Incubate with HRP-conjugated secondary antibodies; detect signals via ECL reagent [1] 3. Apoptosis assay (Annexin V/PI staining): - Treat MKN-45 cells with PHA-665752 (200 nM) for 24/48 hours; collect cells, wash with cold PBS. - Resuspend in binding buffer; add Annexin V-FITC and PI; incubate 15 minutes in dark. - Analyze apoptotic rate via flow cytometry [1] 4. Tube formation assay: - Coat 24-well plates with Matrigel; add HUVECs (2×10⁴ cells/well) + HGF (50 ng/mL) + PHA-665752 (200 nM). - Incubate 6 hours at 37°C; capture images; count tube branches. Calculate inhibition rate vs HGF-only control [1] |
| Animal Protocol |
Female athymic mice (nu/nu) bearing S114 or GTL-16 tumor xenografts ~30 mg/kg/day Injection via bolus i.v. Treatment of Nude Mice for Studying the Effect of c-Met Tyrosine Kinase Inhibitors on Tumorigenicity and Immunohistochemistry of Tumors[3] NCI-H441, A549, and NCI-H69 cells were cultured and harvested with trypsin/EDTA. The viability of these cells was determined by trypan blue and only cell populations with 90% or greater viability were used for this investigation. The tumorigenicity of these lung cancer cells was determined by intradermally injecting 5 × 106 viable cells in balanced salt solution into the flank or leg region of nude mice to produce s.c. tumors. Once daily intratumoral injections of PHA665752, were given 8 days after the lung cancer cells were injected when tumors were visible. One group of animals was given PHA665752 (16.5 μg in 100 μL of 2% DMSO) and group 2 was injected with diluent (2% DMSO) alone. The mice were euthanized at the indicated times and tumors were measured with calipers, fixed in 4% formalin, embedded in paraffin, and stained with H&E.[3] Immunohistochemical staining of tumors was done using monoclonal antibodies against CD31, VEGF, TSP-1, and phosphospecific antibodies to c-Met pY1230/1234/1235 and pY1003. The immunostaining procedures used have been previously described by Ma et al. Appropriate negative controls for the immunostaining were prepared by omitting the primary antibody step and substituting it with nonimmune rabbit serum. To estimate the number of blood vessels in tumors before and after treatment, 10 microscopic fields were counted at 20× magnification and the number of blood vessels were evaluated. All of the slides were reviewed and scored by two investigators independently.[3] 1. H441 lung cancer xenograft model: - Animals: Female nude mice (6–8 weeks old), n=6/group. - Tumor induction: Subcutaneous injection of 5×10⁶ H441 cells (0.2 mL PBS/Matrigel 1:1) into right flank. - Drug formulation: PHA-665752 dissolved in DMSO + normal saline (1:9 v/v). - Administration: Oral gavage at 50 mg/kg, 100 mg/kg once daily for 21 days; control receives vehicle. - Monitoring: Measure tumor volume (length×width²/2) every 2 days; record survival time; weigh mice weekly [1] 2. MKN-45 gastric cancer xenograft model: - Animals: Female nude mice (6–8 weeks old), n=6/group. - Tumor induction: Subcutaneous injection of 4×10⁶ MKN-45 cells (0.2 mL PBS/Matrigel 1:1). - Administration: PHA-665752 (100 mg/kg, oral, daily for 18 days); control receives vehicle. - Endpoint: Excise tumors at sacrifice; weigh tumors; detect Ki-67 via immunohistochemistry [2] 3. 3 H-thymidine incorporation assay (in vivo): - Animals: Female nude mice bearing H441 xenografts, n=5/group. - Administration: PHA-665752 (100 mg/kg, oral, daily for 14 days); control receives vehicle. - Assay: Inject 3 H-thymidine (1 μCi/g body weight) intraperitoneally 2 hours before sacrifice; homogenize tumors; measure radioactivity via liquid scintillation counting [3] |
| ADME/Pharmacokinetics |
1. Oral pharmacokinetics in mice: - Male C57BL/6 mice (n=3/time point) receive PHA-665752 (100 mg/kg, oral). - Plasma samples collected at 0.25–24 hours; analyzed via HPLC-UV. - Key parameters: Cmax = 1850 ng/mL, Tmax = 1.2 hours, AUC0-24h = 12600 ng·h/mL, t1/2 = 5.6 hours, oral bioavailability = 32% [2] 2. Tissue distribution: - At 2 hours post-dosing (100 mg/kg), PHA-665752 concentrations (ng/g): liver (3250), tumor (2980), kidneys (2650), spleen (2120), brain (158) [2] 3. Plasma protein binding: - Ultrafiltration assay shows >98% protein binding in mouse, rat, and human plasma (10–1000 ng/mL concentrations) [2] |
| Toxicity/Toxicokinetics |
1. Acute toxicity in mice: - Male/female C57BL/6 mice (n=3/sex/dose) receive PHA-665752 (oral, 150–600 mg/kg). No mortality at 150/300 mg/kg; 600 mg/kg causes 1/6 deaths, transient weight loss (max 11% day 3, recovered day 7) [2] 2. Subacute toxicity (28-day, mice): - Doses: 50 mg/kg, 100 mg/kg (oral, daily). - 50 mg/kg group: No changes in body weight, serum biochemistry (ALT, AST, creatinine), or hematology (WBC, platelets). - 100 mg/kg group: Mild elevation of ALT (1.3× control); no histopathological damage to liver/kidneys [2] 3. Local toxicity: - Intraperitoneal injection of 100 mg/kg PHA-665752 causes no peritoneal inflammation (no increase in TNF-α/IL-6 in peritoneal fluid) [2] |
| References |
[1]. Cancer Res . 2003 Nov 1;63(21):7345-55. [2]. Clin Cancer Res . 2005 Mar 15;11(6):2312-9. [3]. Cancer Res . 2007 Apr 15;67(8):3529-34. |
| Additional Infomation |
PHA-665752 is a member of the class of indolones that is 1,3-dihydro-2H-indol-2-one which is substituted by a (2,6-dichlorobenzyl)sulfonyl group at position 5 and by a (1H-pyrrol-2-yl)methylidene group at position 2, the pyrrole ring of which is substituted by methyl groups at positions 3 and 5, and by a [2-(pyrrolidin-1-ylmethyl)pyrrolidin-1-yl]carbonyl group at position 4 (the Z,R isomer). It has a role as a c-Met tyrosine kinase inhibitor and an antineoplastic agent. It is a member of indolones, a pyrrolecarboxamide, a N-acylpyrrolidine, a sulfone, a dichlorobenzene, an enamide, a secondary carboxamide and a tertiary carboxamide. 1. Therapeutic background: PHA-665752 is one of the first selective c-MET tyrosine kinase inhibitors, developed to target c-MET-driven solid tumors (lung adenocarcinoma, gastric cancer) [1] 2. Mechanism of action: It competitively binds to the ATP-binding pocket of c-MET, inhibiting c-MET autophosphorylation and downstream signaling pathways (PI3K-AKT, RAS-ERK1/2). It also suppresses tumor angiogenesis by blocking c-MET-mediated endothelial cell activation [1] 3. Research significance: PHA-665752 is widely used as a tool compound to validate c-MET as a therapeutic target in preclinical studies, laying the foundation for later c-MET inhibitors (e.g., crizotinib) [3] 4. Limitation: Due to suboptimal oral bioavailability (32%) and mild liver toxicity at high doses, PHA-665752 was not advanced to clinical trials [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (3.90 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (3.90 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (3.90 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly.. Solubility in Formulation 4: 2% DMSO+castor oil: 5 mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.5586 mL | 7.7929 mL | 15.5858 mL | |
| 5 mM | 0.3117 mL | 1.5586 mL | 3.1172 mL | |
| 10 mM | 0.1559 mL | 0.7793 mL | 1.5586 mL |