PFK-015 (also known as PFK15) is a novel, potent and selective 6-phosphofructo-2-kinase (PFKFB3) inhibitor with IC50 of 207 nM. PFK15 demonstrated cytotoxic effects on Jurkat and H522 lung adenocarcinoma cell lines, with IC50 values of 2.42 and 0.72 μM in Jurkat and H522, respectively. Moreover, PFK15 decreased intracellular ATP, glucose uptake, and F26BP, PFKFB3's substrate. PFK15 (3 μM) increased the number of cells undergoing early apoptosis in Jurkat T cell leukemia cells. In a dose-dependent manner, PFK15 (3 and 20 μM) increased the number of cells going through late apoptosis.
Physicochemical Properties
| Molecular Formula | C17H12N2O | |
| Molecular Weight | 260.29 | |
| Exact Mass | 260.094 | |
| Elemental Analysis | C, 78.44; H, 4.65; N, 10.76; O, 6.15 | |
| CAS # | 4382-63-2 | |
| Related CAS # |
|
|
| PubChem CID | 25142799 | |
| Appearance | Light yellow to yellow solid powder | |
| Density | 1.2±0.1 g/cm3 | |
| Boiling Point | 464.4±45.0 °C at 760 mmHg | |
| Flash Point | 233.5±35.1 °C | |
| Vapour Pressure | 0.0±1.1 mmHg at 25°C | |
| Index of Refraction | 1.698 | |
| LogP | 2.62 | |
| Hydrogen Bond Donor Count | 0 | |
| Hydrogen Bond Acceptor Count | 3 | |
| Rotatable Bond Count | 3 | |
| Heavy Atom Count | 20 | |
| Complexity | 360 | |
| Defined Atom Stereocenter Count | 0 | |
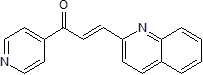
| SMILES | O=C(/C(/[H])=C(\[H])/C1C([H])=C([H])C2=C([H])C([H])=C([H])C([H])=C2N=1)C1C([H])=C([H])N=C([H])C=1[H] |
|
| InChi Key | UJJUKZPBUMCSJZ-BQYQJAHWSA-N | |
| InChi Code | InChI=1S/C17H12N2O/c20-17(14-9-11-18-12-10-14)8-7-15-6-5-13-3-1-2-4-16(13)19-15/h1-12H/b8-7+ | |
| Chemical Name | (E)-1-pyridin-4-yl-3-quinolin-2-ylprop-2-en-1-one | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | recombinant PFKFB3 (IC50 = 110 nM); PFKFB3 activity in cancer cells (IC50 = 20 nM) | |
| ln Vitro |
|
|
| ln Vivo |
PFK15 exhibits sufficient pharmacokinetic characteristics in vivo. In syngeneic mice, PFK15 (25 mg/kg i.p.) inhibits the growth, metastasis, and glucose metabolism of LLC tumors. Comparable to approved chemotherapeutic agents, PFK15 also exhibits antitumor effects in three human xenograft models of cancer in athymic mice. PFK15 Suppresses the Growth of Lewis Lung Carcinoma (LLC) Xenografts [1] Intraperitoneal administration of 25 mg/kg PFK15 every three days caused <10% body mass loss and no end organ toxicity based on blood, urine and histological analyses. Since 25 mg/kg of PFK15 was easily administered in a small volume despite the limited solubility of PFK15, this non-toxic dose and schedule was selected for the efficacy testing in xenograft and syngeneic models of tumorigenesis. We initially examined the relative effects of daily intraperitoneal administration of PFK15 on the growth of established LLC tumors in vivo. Although we did not observe tumor regression as a result of PFK15 administration, we did observe a statistically significant reduction in the growth of the LLC tumors (Fig. 4A) without a major effect on body mass (Fig. 4B). Importantly, whereas LLC cells are well established to metastasize from the subcutaneous tumors to the lungs, no lung metastases were identified in the LLC-bearing mice that had been treated with PFK15 (Fig. 4C).We then examined a subset of tumors for F26BP and apoptotic cells four days after initiation of PFKFB3 inhibitor administration (n=4). We found that PFK15 reduced tumor-associated F26BP (Fig. 4D) and caused a marked increase in the number of cells that were positive for cleaved caspase 3 by immunohistochemistry (Fig. 4E and F). Taken together, these results suggest that PFK15 can reduce tumor growth and induce apoptosis in vivo, is well tolerated in terms of body mass and has the unanticipated ability to prevent either the initiation or growth of metastases. PFK15 Suppresses 18F-FDG Uptake by Lewis Lung Carcinoma (LLC) Xenografts [1] We postulated that PFK15 would acutely attenuate the glucose uptake of xenografted tumors in vivo. We conducted baseline 18F-FDG-PET imaging of LLC-bearing C57Bl/6 mice and, 24 hours later, injected PFK15 and then reimaged the LLC-bearing mice after 45 minutes. We observed a ~50% reduction in 18F-FDG uptake after PFK15 administration normalized to the FDG uptake in the cerebellum (neurons do not express PFKFB3 protein; Fig. 4G and H). These data demonstrate that PFK15 selectively suppresses glucose uptake by tumors in vivo and suggest that FDG uptake scans may be a useful pharmacodynamic endpoint of PFKFB3 inhibitors in clinical trials. PFK15 Displays Anti-Tumor Activity That Is Similar to Commonly Used Chemotherapeutic Agents [1] Last, we directly compared the anti-tumor effects of PFK15 to three chemotherapeutic agents that are FDA-approved for the treatment of human cancers. We found that PFK15 suppressed the growth of colon and pancreatic adenocarcinomas similarly to irinotecan (Fig. 5A) and gemcitabine (Fig. 5C), respectively. However, the activity of PFK15 against glioblastoma growth was lower than that observed by temozolomide (Fig. 5B). These pre-clinical studies raise the prospect that PFK15 and close synthetic derivatives may demonstrate cytotoxic activity against human cancers in clinical trials. |
|
| Enzyme Assay |
For kinase reactions, 13 ng of recombinant human PFKFB3 protein is incubated for 1 hour at room temperature in a reaction mix that contains 10 μmol/L ATP, 10 μmol/L F6P, and either dimethyl sulfoxide (DMSO) vehicle control, 3PO, or PFK15. According to the manufacturer, kinase activity is determined using the Adapta Universal Kinase Assay.
Recombinant PFKFB3 Assay [1] Kinase reactions were performed by incubating 13ng of recombinant human PFKFB3 protein in a reaction mix containing 10μM ATP, 10μM F6P, and either DMSO vehicle control, 3PO, or PFK15 for one hour at room temperature. Kinase activity was measured with the Adapta Universal Kinase Assay per manufacturer’s instructions and fluorescent and FRET signals were measured on a Tecan Safire2 plate reader. Graphs were generated using SigmaPlot software and IC50 values were calculated using a three parameter Hill Equation. The KINOMEscanEDGE list of 96 kinases can be accessed at http://www.kinomescan.com. |
|
| Cell Assay |
2-[1-14C]-deoxy-D-glucose uptake [1] Jurkat or H522 cells were plated in RPMI 1640 media supplemented with 10% FCS and 50μg/ml gentamycin at 100,000/ml and immediately treated with DMSO vehicle control, 3PO, or PFK-15 for three hours. Cells were washed twice with pre-warmed glucose free RPMI media and incubated for 30 minutes in the presence of the PFKFB3 inhibitors. Cells were treated with 25μl of 14C-deoxy-glucose (0.1μCi/μl) for one hour, washed once with ice-cold glucose free RPMI and twice with ice cold PBS. Cells were lysed in 500μl 0.5% SDS. 400μl of lysate was added to 5ml Microscint 40 scintillation fluid and counts were measured on a Tri-Carb 2910 liquid scintillation analyzer. Remaining lysate was quantitated with the BCA Assay per manufacturer’s instructions and protein levels measured on a Powerwave XS plate reader. Counts were normalized to protein concentration. ATP assay [1] Cells were washed (while still adherent) with cold PBS ×1, lysed with passive lysis buffer (1X) added directly to the plates and immediately harvested by scraping. The lysates were flash frozen (to −80°C) and thawed (to 37°C) once to accomplish complete lysis and then centrifuged (at 4°C) for 30 seconds to clear the lysates. Intracellular ATP levels were determined using a bioluminescence assay utilizing recombinant firefly luciferase and its substrate, D-luciferin. The luminescence was read in a TD-20/20 luminometer at 560 nm. The ATP values were calculated using an ATP standard curve. The protein concentrations of the lysates were estimated using the BCA assay and ATP was expressed as nmol per mg protein. Flow Cytometry [1] Jurkat cells were plated in RPMI 1640 media supplemented with 10% FCS and 50μg/ml gentamycin at 100,000 cells/ml and immediately incubated with DMSO vehicle control, 3PO, PFK-15, or Etoposide for five hours. Cells were washed with PBS and stained with Annexin V and Propidium Iodide. Fluorescence was measured using a FACSCalibur and analyzed using FloJo.Annexin V+/PI+ (late apoptotic) and Annexin V+/PI− (early apoptotic) cells were quantified by the frequency of fluorescently labeled cells and statistical significance was assessed by the two-sample T-test (independent variable). Trypan blue exclusion is used to determine viability. For five minutes, the cells were incubated in 20% trypan blue. A standard hemocytometer is used to count all of the viable cells, excluding trypan blue cells. There are three duplicates of each experiment. |
|
| Animal Protocol |
C57Bl/6 mice bearing LLC xenografts, Balb/C athymic mice bearing CT26, U-87 MG, or BxPC-3 xenografts. 25 mg/kg every 3 days i.p. Pharmacokinetic Studies [1] The PK profile was determined in female C57Bl/6 mice following IV administration of the PFKFB3 inhibitors. Using only female mice lowered the number of animals required for meaningful results without the issues of potential gender differences in exposure. Eight time points and three animals per time point were used to determine the PK parameters calculated using WinNonLin v5.0 (Tmax, Cmax, volume of distribution, half-life, clearance, AUCs and MRT). Plasma samples were extracted using acetonitrile and analyzed by LC/MS-MS using a PhenomexSynergi Polar-RP 4micron 50×2.0 mm column eluted with a biphasic mobile phase (0.5% formic acid in acetonitrile and water). FDG-PET Imaging [1] Tumor-bearing mice were anesthetized with isoflurane and 200 μCi of FDG was administered intravenously via the tail veins. The baseline micro-PET imaging of FDG in the mice was acquired by the ordered subsets expectation maximization (OSEM), using 6 subsets/4 iterations for 15-minute static scans 45 minutes after IV injection of the tracer using a Concorde Microsystems 4-ring micro-PET system. Regions of interest in the tumor and cerebellum were quantified in quadruplicate and, 24 hours later (18F half-life, 109.8 min), PFK-15 (25 mg/kg) was administered i.p. and the micro-PET scan was repeated 45 minutes later. Xenograft Studies [1] LLC, CT26, U-87 or BxPC-3 cells were collected from exponential growth phase culture in DMEM supplemented with 10% FCS. Cells were washed twice and re-suspended in PBS (1×107 cells/ml). Groups of C57Bl6 (for LLC cells) or Balb/C athymic mice(for CT26, U-87 MG or BxPC-3 cells) (20 gm) were injected s.c. with 0.1 ml of the cell suspension (1 ×106 cells). Tumor masses were determined in a blinded fashion with Vernier calipers according to the following formula: mass(mg)=(width, mm)2×(length, mm)/2 (19)and then monitored daily with microcalipers. Mice bearing xenografts (150–200 mg) were then randomized to DMSO, PFK-15 (25 mg/kg i.p. every 3 days × 4), irinotecan (70 mg/kg i.v. every 3 days × 4), temozolomide (30 mg/kg p.o. 3 days on 3 days off), or gemcitabine (100 mg/kg every 3 days × 4) and microcaliper measurements were conducted daily. All data are expressed as the mean±SD of two experiments (n = 8 per group). Statistical significance was assessed by the unpaired two tail T-test. In a separate series of experiments, LLC-tumor bearing mice were euthanized after only four days of treatment and the tumors were excised, fixed in formalin, embedded in paraffin and stained with Hematoxylin and Eosin or with anti-cleaved caspase 3 using standard immunohistochemical methodologies. |
|
| References |
[1]. Mol Cancer Ther . 2013 Aug;12(8):1461-70. |
|
| Additional Infomation |
In human cancers, loss of PTEN, stabilization of hypoxia inducible factor-1α, and activation of Ras and AKT converge to increase the activity of a key regulator of glycolysis, 6-phosphofructo-2-kinase (PFKFB3). This enzyme synthesizes fructose 2,6-bisphosphate (F26BP), which is an activator of 6-phosphofructo-1-kinase, a key step of glycolysis. Previously, a weak competitive inhibitor of PFKFB3, 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO), was found to reduce the glucose metabolism and proliferation of cancer cells. We have synthesized 73 derivatives of 3PO and screened each compound for activity against recombinant PFKFB3. One small molecule, 1-(4-pyridinyl)-3-(2-quinolinyl)-2-propen-1-one (PFK15), was selected for further preclinical evaluation of its pharmacokinetic, antimetabolic, and antineoplastic properties in vitro and in vivo. We found that PFK15 causes a rapid induction of apoptosis in transformed cells, has adequate pharmacokinetic properties, suppresses the glucose uptake and growth of Lewis lung carcinomas in syngeneic mice, and yields antitumor effects in three human xenograft models of cancer in athymic mice that are comparable to U.S. Food and Drug Administration-approved chemotherapeutic agents. As a result of this study, a synthetic derivative and formulation of PFK15 has undergone investigational new drug (IND)-enabling toxicology and safety studies. A phase I clinical trial of its efficacy in advanced cancer patients will initiate in 2013 and we anticipate that this new class of antimetabolic agents will yield acceptable therapeutic indices and prove to be synergistic with agents that disrupt neoplastic signaling.[1] Metabolic inhibition via PFKFB3 inhibition has demonstrated considerable tumor inhibitory effects in various studies; however, PFKFB3 inhibition did not show satisfactory tumor inhibition when used in clinical trials. PFKFB3 is a crucial metabolic enzyme that is highly upregulated in cancer cells and directly affects tumor glycolysis. Here, we showed that PFKFB3 inhibition suppresses tumors in vitro and in vivo in immune-deficient xenografts. However, this inhibition induces the upregulation of PD-L1 levels, which inactivated cocultured T-cells in vitro, compromises anti-tumor immunity in vivo, and reduced anti-tumor efficacy in an immune-competent mouse model. Functionally, PD-1 mAb treatment enhances the efficacy of PFKFB3 inhibition in immunocompetent and hu-PBMC NOG mouse models. Mechanistically, PFKFB3 inhibition increases phosphorylation of PFKFB3 at residue Ser461, which increases interaction with HIF-1α, and their colocalization into the nucleus, where HIF-1α transcriptionally upregulate PD-L1 expression and causes subsequent tumor immune evasion. Higher phos-PFKFB3 correlated with higher PD-L1 expression, lower CD8 and GRZMB levels, and shorter survival time in ESCC patients.Oncoimmunology. 2022 May 25;11(1):2079182. |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2 mg/mL (7.68 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2 mg/mL (7.68 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2 mg/mL (7.68 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly.. Solubility in Formulation 4: 5% DMSO +45% PEG 300 +1% Tween 80 +ddH2O: 5mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.8419 mL | 19.2093 mL | 38.4187 mL | |
| 5 mM | 0.7684 mL | 3.8419 mL | 7.6837 mL | |
| 10 mM | 0.3842 mL | 1.9209 mL | 3.8419 mL |