PF-4708671 is a novel, potent, highly specific and cell-permeable inhibitor of p70 ribosomal S6 kinase (S6K1 isoform)/RSK1 with potential antitumor activity. In cell-free assays, it inhibits RSK1 with a Ki/IC50 of 20 nM/160 nM, shows 400-fold greater selectivity for S6K1 over S6K2, and exhibits 4- and >20-fold selectivity for S6K1 over MSK1 and RSK1/2, respectively. As the first S6K1-specific inhibitor reported, PF-4708671 showed potent antiproliferative activity in vitro and high antitumor efficacy in vivo by preventing the S6K1-mediated phosphorylation of S6 protein in response to IGF-1 (insulin-like growth factor 1), while having no effect upon the PMA-induced phosphorylation of substrates of the highly related RSK (p90 ribosomal S6 kinase) and MSK (mitogen- and stress-activated kinase) kinases.
Physicochemical Properties
| Molecular Formula | C19H21N6F3 |
| Molecular Weight | 390.40544 |
| Exact Mass | 390.177 |
| Elemental Analysis | C, 58.45; H, 5.42; F, 14.60; N, 21.53 |
| CAS # | 1255517-76-0 |
| Related CAS # | 1255517-76-0 |
| PubChem CID | 51371303 |
| Appearance | White to off-white solid powder |
| Density | 1.3±0.1 g/cm3 |
| Boiling Point | 572.8±50.0 °C at 760 mmHg |
| Flash Point | 300.2±30.1 °C |
| Vapour Pressure | 0.0±1.6 mmHg at 25°C |
| Index of Refraction | 1.609 |
| LogP | 3.2 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 8 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 28 |
| Complexity | 510 |
| Defined Atom Stereocenter Count | 0 |
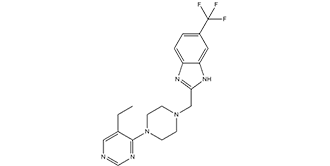
| SMILES | FC(C1=CC=C2N=C(CN3CCN(C4=NC=NC=C4CC)CC3)NC2=C1)(F)F |
| InChi Key | FBLPQCAQRNSVHB-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C19H21F3N6/c1-2-13-10-23-12-24-18(13)28-7-5-27(6-8-28)11-17-25-15-4-3-14(19(20,21)22)9-16(15)26-17/h3-4,9-10,12H,2,5-8,11H2,1H3,(H,25,26) |
| Chemical Name | 2-((4-(5-ethylpyrimidin-4-yl)piperazin-1-yl)methyl)-5-(trifluoromethyl)-1H-benzo[d]imidazole |
| Synonyms | PF-4708671; PF4708671; PF 4708671 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
S6K1 (IC50 = 160 nM); S6K1 (Ki = 20 nM); S6K2 (IC50 = 65 μM) The target of PF-4708671 is p70 ribosomal S6 kinase 1 (S6K1). For recombinant human S6K1, its half-maximal inhibitory concentration (IC50) is approximately 20 nM. It shows low cross-reactivity with S6K2 (a homologous isoform of S6K1) with an IC50 of ~360 nM, and no significant inhibitory activity against other kinases including Akt1 (IC50 > 10 μM), ERK2 (IC50 > 10 μM), mTOR (IC50 > 10 μM), and PKCα (IC50 > 10 μM), demonstrating high selectivity for S6K1 [1] PF-4708671 maintains specific inhibition of S6K1 in colon carcinoma cells, with no detected off-target effects on the insulin-like growth factor 1 receptor (IGF-1R) or its downstream kinases other than S6K1 [2] |
| ln Vitro |
PF-4708671 inhibits the activity of full-length S6K1 in vitro with a Ki of 20 nM, and S6K1 isolated from IGF1-stimulated HEK293 cells with an IC50 of 0.16 μM, and only very slightly (IC50 of 65 μM) inhibits the closely related S6K2 isoform. Over 20 times less potently than S6K1, PF-4708671 inhibits RSK1 (IC50 of 4.7 μM) and RSK2 (IC50 of 9.2 μM). PF4708671 inhibits MSK1 four times less potently than S6K1 (IC50 = 0.95 μM)[1]. HCT116 cells are treated with (i) vehicle (DMSO), (ii) OSI-906 (5 μM), (iii) PF-4708671 (10 μM), and (iv) OSI-906 (5 μM)+PF-4708671 (10 μM) for various amounts of time. OSI-906 alone (closed square) or PF4708671 alone (open circle) treatments on HCT116 cells marginally inhibit cell growth. The combination of OSI-906 and PF-4708671, on the other hand, significantly reduces HCT116 cell proliferation after a 2-day treatment (closed circle). When SW480 cells are treated with the mixture of OSI-906 and PF-4708671, the outcome is also similar. In comparison to vehicle, OSI-906 alone, or PF-4708671 alone treated HCT116 or SW480 cells, colony formation is also significantly decreased in OSI-906+PF-4708671-treated cells[2]. 1. S6K1 enzyme activity inhibition (from [1]): In vitro assays using purified recombinant human S6K1 showed that PF-4708671 dose-dependently inhibited S6K1-mediated phosphorylation of its specific substrate peptide (sequence: RRRLSSLRA). The inhibition curve fitted from 7 concentration gradients (0.1 nM to 1000 nM) confirmed an IC50 of ~20 nM, which is consistent with its target binding specificity [1] 2. Inhibition of S6K1 downstream signaling (from [1]): Treatment of HEK293 cells with PF-4708671 (0.2 μM, 1 μM, 5 μM) for 4 hours resulted in a concentration-dependent reduction in the phosphorylation levels of S6K1 (at Thr389) and its downstream substrate S6 (at Ser235/236), as detected by Western blot. Specifically, 1 μM PF-4708671 inhibited ~80% of phosphorylated S6K1 (p-S6K1, Thr389) and ~75% of phosphorylated S6 (p-S6, Ser235/236), while the total protein levels of S6K1 and S6 remained unchanged. Similar results were observed in MCF-7 breast cancer cells and HCT116 colon cancer cells [1] 3. Antiproliferative activity (from [1]): Using the MTT assay to evaluate cell viability, PF-4708671 exhibited dose-dependent antiproliferative effects on multiple human cancer cell lines. For MCF-7 breast cancer cells, the IC50 for 72-hour proliferation inhibition was ~1.2 μM; for HCT116 colon cancer cells, the IC50 was ~1.8 μM; for HeLa cervical cancer cells, the IC50 was ~2.1 μM. No significant antiproliferative effect was observed on normal human foreskin fibroblasts (NHFF) even at concentrations up to 10 μM [1] 4. Overcoming IGF-1R inhibitor resistance (from [2]): In IGF-1R inhibitor-resistant colon cancer cells (HCT116/IGF-1Ri), treatment with PF-4708671 alone (0.5 μM, 1 μM, 2 μM, 4 μM) for 72 hours inhibited cell viability in a dose-dependent manner, with an IC50 of ~1.7 μM (comparable to the parental HCT116 cells’ IC50 of ~1.5 μM). When combined with the IGF-1R inhibitor NVP-AEW541 (1 μM), PF-4708671 (2 μM) significantly enhanced the inhibitory effect on HCT116/IGF-1Ri cell viability: the viability rate decreased from 65% (NVP-AEW541 alone) to 28% (combination treatment). Additionally, the combination reduced the number of colonies formed by HCT116/IGF-1Ri cells by ~60% (vs. NVP-AEW541 alone) and increased the apoptotic rate (Annexin V-positive cells) from 12% (vehicle control) to 35% (combination treatment) [2] |
| ln Vivo |
The tumor growth rate in mice treated with the combination of OSI-906+PF-4708671 is significantly slower than that of OSI-906 alone (P=0.0189) or PF4708671 alone (P=0.0165) treated mice. At the conclusion of a 15-day treatment period, the average tumor volume in OSI-906+PF-4708671-treated mice is approximately 50% smaller than that in mice treated with OSI-906 (P=0.0056) or PF-4708671 alone (P<0.001)[2]. 1. Antitumor efficacy in HCT116 xenograft model (from [1]): Female nude mice (6–8 weeks old) were subcutaneously inoculated with HCT116 colon cancer cells (5×10⁶ cells/mouse) in the right flank. When tumors reached an average volume of 100 mm³, mice were randomly divided into 3 groups (n=6/group): (1) Vehicle control (10% DMSO + 10% Tween 80 + 80% normal saline, administered via oral gavage); (2) PF-4708671 50 mg/kg (dissolved in the same vehicle as control, oral gavage, once daily); (3) PF-4708671 100 mg/kg (same vehicle and administration route/frequency as the 50 mg/kg group). Treatment lasted for 21 days, with tumor volume measured every 3 days using calipers (tumor volume = length × width² / 2). At the end of treatment, the 100 mg/kg group showed a significant tumor growth inhibition (TGI) rate of ~45% (average tumor volume: 480 ± 52 mm³ vs. 870 ± 65 mm³ in the control group). No significant changes in body weight (average weight loss < 5%) or signs of toxicity (e.g., lethargy, diarrhea, hair loss) were observed in either drug group. Western blot analysis of tumor tissues from the 100 mg/kg group revealed a ~60% reduction in p-S6K1 (Thr389) and p-S6 (Ser235/236) levels compared to the control group [1] |
| Enzyme Assay |
PF-4708671 is a cell-permeable inhibitor of p70 ribosomal S6 kinase (S6K1 isoform) with Ki/IC50 of 20 nM/160 nM in cell-free assays, 400-fold greater selectivity for S6K1 than S6K2, and 4- and >20-fold selectivity for S6K1 than MSK1 and RSK1/2, respectively. 1. S6K1 kinase activity assay (from [1]): - Reagent preparation: Purified recombinant human S6K1 catalytic domain (residues 224–502) was prepared first. The specific S6K1 substrate peptide (sequence: RRRLSSLRA) was dissolved in distilled water to a stock concentration of 1 mM. [γ-³²P]ATP (specific activity ~3000 Ci/mmol) was diluted with non-radioactive ATP to a final concentration of 10 μM (radioactive:non-radioactive ratio = 1:100). A reaction buffer containing 50 mM Tris-HCl (pH 7.5), 10 mM MgCl₂, 1 mM dithiothreitol (DTT), and 0.1 mg/mL bovine serum albumin (BSA) was formulated [1] - Assay setup: PF-4708671 was serially diluted in dimethyl sulfoxide (DMSO) to obtain 7 concentration gradients (0.1 nM, 0.3 nM, 1 nM, 3 nM, 10 nM, 30 nM, 1000 nM). Each dilution was added to the reaction mixture (final DMSO concentration ≤ 1%, which had no effect on S6K1 activity). The reaction mixture contained reaction buffer, substrate peptide (final concentration 100 μM), and [γ-³²P]ATP (final concentration 10 μM). Recombinant S6K1 (final concentration 5 nM) was added to initiate the reaction, and the mixture was incubated at 30°C for 30 minutes. A vehicle control (DMSO only) and a positive control (a known non-selective S6K inhibitor) were included, with 3 technical replicates per group [1] - Detection and analysis: After incubation, 20 μL of each reaction mixture was spotted onto phosphocellulose filter papers (P81 type). The filters were washed 3 times with 1% phosphoric acid (5 minutes per wash) to remove unincorporated [γ-³²P]ATP, rinsed once with acetone to remove residual moisture, and air-dried at room temperature. The radioactivity on the filters was measured using a liquid scintillation counter. The inhibition rate of S6K1 activity was calculated as [(Radioactivity of vehicle control - Radioactivity of sample) / Radioactivity of vehicle control] × 100%. The IC50 value was determined by fitting the inhibition rates of different PF-4708671 concentrations to a four-parameter logistic regression model using graphing software [1] |
| Cell Assay |
These cells include GEO, HT29, SW480, and HCT116. XTT and clonogenic assays are used to determine how OSI-906 or OSI-906 and PF-4708671 affect cell proliferation. Cell Proliferation Kit II (XTT) is used to carry out XTT assays. For clonogenic assays, cells (1 103 cells/well) are seeded on a 6-well plate and then given drug treatments (OSI-906 5 M, PF-4708671 10 M). Cells are stained with 1% crystal violet after one week of incubation, and the number of colonies is counted and noted[2]. 1. Antiproliferation assay (MTT method, from [1]): - Cell seeding: Human cancer cell lines (HCT116, MCF-7, HeLa) and normal NHFF cells were trypsinized and resuspended in complete medium (DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin). Cells were seeded into 96-well plates at a density of 5×10³ cells per well and incubated at 37°C with 5% CO₂ for 24 hours to allow cell attachment [1] - Drug treatment: PF-4708671 was diluted with complete medium to 8 concentration gradients (0.1 μM, 0.3 μM, 1 μM, 3 μM, 10 μM, 30 μM, 100 μM) (final DMSO concentration ≤ 1%). The original medium in each well was aspirated and replaced with 100 μL of the diluted PF-4708671 solution. A vehicle control group (complete medium containing 1% DMSO) was set up. Each concentration was tested in 3 biological replicates, and the plates were incubated at 37°C with 5% CO₂ for 72 hours [1] - Viability detection: After incubation, 20 μL of MTT solution (5 mg/mL in phosphate-buffered saline, PBS) was added to each well, and the plates were incubated for another 4 hours. The supernatant was carefully aspirated to avoid disturbing the formazan crystals formed at the bottom of the wells. 150 μL of DMSO was added to each well to dissolve the crystals, and the plates were shaken gently for 10 minutes at room temperature. The absorbance at 570 nm was measured using a microplate reader. The cell viability percentage was calculated as (Absorbance of sample well / Absorbance of vehicle control well) × 100%, and the IC50 for antiproliferation was obtained by fitting the viability data to a logistic regression model [1] 2. Western blot assay for S6K1 downstream signaling (from [1]): - Cell preparation and treatment: HEK293, MCF-7, or HCT116 cells were seeded into 6-well plates at a density of 2×10⁵ cells per well and cultured at 37°C with 5% CO₂ until reaching 70–80% confluence. The medium was replaced with fresh complete medium containing PF-4708671 at concentrations of 0.2 μM, 1 μM, and 5 μM (vehicle control: complete medium with 1% DMSO). The cells were incubated for 4 hours [1] - Protein extraction and electrophoresis: Cells were washed twice with ice-cold PBS to remove residual medium. 200 μL of RIPA lysis buffer (supplemented with protease and phosphatase inhibitors) was added to each well, and the cells were scraped off on ice. The lysates were transferred to microcentrifuge tubes and centrifuged at 12,000 × g for 15 minutes at 4°C to collect the supernatant (total cellular protein). The protein concentration was determined using a BCA protein assay kit. Equal amounts of protein (30 μg per lane) were mixed with 5× SDS loading buffer, heated at 95°C for 5 minutes to denature, and separated by 10% SDS-PAGE. The separated proteins were transferred to a PVDF membrane using a wet transfer system [1] - Immunodetection: The PVDF membrane was blocked with 5% non-fat milk in TBST buffer (20 mM Tris-HCl, 150 mM NaCl, 0.1% Tween 20) for 1 hour at room temperature. The membrane was then incubated overnight at 4°C with primary antibodies against p-S6K1 (Thr389), total S6K1, p-S6 (Ser235/236), total S6, and β-actin (loading control). After incubation, the membrane was washed 3 times with TBST (5 minutes per wash) and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 hour at room temperature. The membrane was washed again 3 times with TBST, and the protein bands were visualized using an enhanced chemiluminescence (ECL) reagent. The band intensity was quantified using image analysis software, and the ratio of phosphorylated protein to total protein was calculated to evaluate the inhibition of S6K1 downstream signaling by PF-4708671 [1] 3. Cell viability and apoptosis assays in resistant colon cancer cells (from [2]): - Cell viability assay: HCT116/IGF-1Ri cells were seeded into 96-well plates at 4×10³ cells per well and incubated for 24 hours. The medium was replaced with medium containing PF-4708671 alone (0.5 μM, 1 μM, 2 μM, 4 μM) or in combination with NVP-AEW541 (1 μM). A vehicle control group and an NVP-AEW541 alone group were set up. After 72 hours of incubation, cell viability was measured using the MTT method (procedure consistent with the antiproliferation assay in [1]), and the viability rate was calculated [2] - Colony formation assay: HCT116/IGF-1Ri cells were seeded into 6-well plates at 500 cells per well and incubated for 24 hours. The medium was replaced with medium containing PF-4708671 (2 μM) + NVP-AEW541 (1 μM), NVP-AEW541 alone (1 μM), or vehicle control. The medium was refreshed every 3 days. After 14 days of incubation, the colonies were fixed with 4% paraformaldehyde for 15 minutes, stained with 0.1% crystal violet for 30 minutes, and rinsed with water to remove excess stain. The number of colonies (≥50 cells per colony) was counted manually, and the colony formation rate was calculated as (Number of colonies in treatment group / Number of colonies in control group) × 100% [2] - Apoptosis assay: HCT116/IGF-1Ri cells were seeded into 6-well plates at 2×10⁵ cells per well and incubated for 24 hours. The cells were treated with PF-4708671 (2 μM) + NVP-AEW541 (1 μM), NVP-AEW541 alone (1 μM), or vehicle control for 48 hours. The cells were harvested by trypsinization, washed twice with cold PBS, and resuspended in binding buffer. Annexin V-FITC and propidium iodide (PI) were added to the cell suspension, and the mixture was incubated in the dark for 15 minutes at room temperature. The apoptotic rate (Annexin V-positive cells) was detected using a flow cytometer [2] |
| Animal Protocol |
Mice: The following groups (five mice/group) of female athymic nude mice (Hsd:Athymic Nude-Foxn1nu) are randomly assigned. Mice are given either OSI-906 (30 mg/kg) or vehicle (25 mM tartaric acid) for 12 days prior to the injection of HT29-L and HT29-P cells. Mice are administered the following drugs orally for 14 days prior to the injection of HCT116 cells: vehicle (25 mM tartaric acid); OSI-906 alone (30 mg/kg); PF-4708671 alone (60 mg/kg); and OSI-906 (30 mg/kg)+PF-4708671 (60 mg/kg). Each day, one dose of OSI-906, one dose of PF-4708671, and one dose of Vehicle are administered. The mice are sacrificed, and the weights of the tumors were measured, twenty-four hours after the last treatment[2]. 1. HCT116 xenograft tumor experiment (from [1]): - Animal selection and housing: Female nude mice (6–8 weeks old, specific pathogen-free grade) were housed in a controlled environment with a 12-hour light/dark cycle, constant temperature (22±2°C), and constant humidity (50±5%). Mice had free access to standard rodent chow and sterile water [1] - Tumor cell inoculation: HCT116 colon cancer cells were cultured to logarithmic growth phase, trypsinized, and resuspended in PBS at a concentration of 5×10⁷ cells/mL. Each mouse was subcutaneously injected with 0.1 mL of the cell suspension (5×10⁶ cells) into the right flank [1] - Grouping and drug administration: When tumors grew to an average volume of 100 mm³, mice were randomly divided into 3 groups (n=6/group). The vehicle control group received 10% DMSO + 10% Tween 80 + 80% normal saline via oral gavage once daily. The low-dose drug group received PF-4708671 50 mg/kg (dissolved in the same vehicle as the control) via oral gavage once daily. The high-dose drug group received PF-4708671 100 mg/kg (same vehicle and administration route/frequency as the low-dose group). The treatment duration was 21 days [1] - Sample collection and detection: During the treatment period, mouse body weight and tumor volume were measured every 3 days. At the end of treatment, mice were euthanized by cervical dislocation. Tumors were excised, weighed, and a portion of each tumor was frozen in liquid nitrogen for subsequent Western blot analysis (to detect p-S6K1 and p-S6 levels), while the remaining portion was fixed in 4% paraformaldehyde for pathological analysis (not related to PF-4708671 specific activity) [1] |
| References |
[1]. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem J. 2010 Oct 15;431(2):245-55. [2]. Inhibition of p70S6K1 Activation by Pdcd4 Overcomes the Resistance to an IGF-1R/IR Inhibitor in Colon Carcinoma Cells. Mol Cancer Ther. 2015 Mar;14(3):799-809. |
| Additional Infomation |
S6K1 (p70 ribosomal S6 kinase 1) is activated by insulin and growth factors via the PI3K (phosphoinositide 3-kinase) and mTOR (mammalian target of rapamycin) signalling pathways. S6K1 regulates numerous processes, such as protein synthesis, growth, proliferation and longevity, and its inhibition has been proposed as a strategy for the treatment of cancer and insulin resistance. In the present paper we describe a novel cell-permeable inhibitor of S6K1, PF-4708671, which specifically inhibits the S6K1 isoform with a Ki of 20 nM and IC50 of 160 nM. PF-4708671 prevents the S6K1-mediated phosphorylation of S6 protein in response to IGF-1 (insulin-like growth factor 1), while having no effect upon the PMA-induced phosphorylation of substrates of the highly related RSK (p90 ribosomal S6 kinase) and MSK (mitogen- and stress-activated kinase) kinases. PF-4708671 was also found to induce phosphorylation of the T-loop and hydrophobic motif of S6K1, an effect that is dependent upon mTORC1 (mTOR complex 1). PF-4708671 is the first S6K1-specific inhibitor to be reported and will be a useful tool for delineating S6K1-specific roles downstream of mTOR.[1] Agents targeting insulin-like growth factor 1 receptor (IGF-1R) are being actively examined in clinical trials. Although there has been some initial success of single-agent targeting IGF-1R, attempts in later studies failed because of resistance. This study aimed to understand the effects of programmed cell death 4 (Pdcd4) on the chemosensitivity of the IGF-1R inhibitor OSI-906 in colorectal cancer cells and the mechanism underlying this impact. Using OSI-906-resistant and -sensitive colorectal cancer cells, we found that the Pdcd4 level directly correlates with cell chemosensitivity to OSI-906. In addition, tumors derived from Pdcd4 knockdown cells resist the growth inhibitory effect of OSI-906 in a colorectal cancer xenograft mouse model. Moreover, Pdcd4 enhances the antiproliferative effect of OSI-906 in resistant cells through suppression of p70S6K1 activation. Knockdown of p70S6K1, but not p70S6K2, significantly increases the chemosensitivity of OSI-906 in cultured colorectal cancer cells. Furthermore, the combination of OSI-906 and PF-4708671, a p70S6K1 inhibitor, efficiently suppresses the growth of OSI-906-resistant colon tumor cells in vitro and in vivo. Taken together, activation of p70S6K1 that is inhibited by Pdcd4 is essential for resistance to the IGF-1R inhibitor in colon tumor cells, and the combinational treatment of OSI-906 and PF-4708671 results in enhanced antiproliferation effects in colorectal cancer cells in vitro and in vivo, providing a novel venue to overcome the resistance to the IGF-1R inhibitor in treating colorectal cancer.[2] 1. Mechanism of action (from [1]): PF-4708671 is a small-molecule inhibitor designed to target the ATP-binding pocket of S6K1. It exerts its inhibitory effect by competing with ATP for binding to the catalytic domain of S6K1, thereby blocking the kinase activity of S6K1 and inhibiting the phosphorylation of its downstream substrates (e.g., S6). This mechanism ultimately suppresses the translation of ribosomal proteins and cell cycle progression, leading to antiproliferative effects on cancer cells [1] 2. Research application as a tool compound (from [1]): Due to its high selectivity for S6K1, PF-4708671 is widely used as a tool compound in basic research to explore the biological functions of S6K1, especially its role in the mTOR signaling pathway, cell proliferation, and cancer progression. It helps validate S6K1 as a potential therapeutic target for cancers with dysregulated mTOR-S6K1 signaling [1] 3. Potential in overcoming drug resistance (from [2]): In IGF-1R inhibitor-resistant colon cancer cells, PF-4708671 can restore sensitivity to IGF-1R inhibitors by inhibiting S6K1 activity. This finding suggests that combining PF-4708671 with IGF-1R inhibitors may be a potential therapeutic strategy for treating IGF-1R inhibitor-resistant colon cancer, though this has not been verified in clinical trials [2] 4. Development status (from [1] and [2]): PF-4708671 is a preclinical research compound and has not entered clinical development. It is primarily used as an experimental tool to study S6K1 function and related signaling pathways, rather than a candidate drug for clinical treatment [1][2] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~30 mg/mL (76.8 mM) Water: <1 mg/mL Ethanol: 8 mg/mL (20.5 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (6.40 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (6.40 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (6.40 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 30% PEG400+0.5% Tween80+5% propylene glycol: 30mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5614 mL | 12.8070 mL | 25.6141 mL | |
| 5 mM | 0.5123 mL | 2.5614 mL | 5.1228 mL | |
| 10 mM | 0.2561 mL | 1.2807 mL | 2.5614 mL |